INTRODUCTION
PRODUCTION AND PROPERTIES OF X-RADIATION
THE DIFFRACTION OF X-RAYS BY CRYSTALLINE POWDERS
THE MEASUREMENT OF INTERPLANAR SPACINGS ( d) AND THE ESTIMATION OF RELATIVE INTENSITIES ( I/I1)
IDENTIFICATION FROM X-RAY DIFFRACTION POWDER DATA
PREPARATION AND SIZE OF SPECIMENS, CHOICE OF RADIATION, AND THE TIME FACTOR
APPRAISAL OF THE X-RAY DIFFRACTION POWDER METHOD FOR NARCOTICS IDENTIFICATION
ACKNOWLEDGEMENTS
Author: W. H. Barnes
Pages: 20 to 31
Creation Date: 1954/01/01
TABLE OF CONTENTS
|
|
Page |
|
Introduction |
20 |
|
Production and properties of X-radiation |
20 |
|
The diffraction of X-rays by crystalline powders |
21 |
|
The measurement of interplanar spacings ( d) and the esti- mation of relative intensities ( I/I 1) |
24 |
|
Identification from X-ray diffraction powder data |
26 |
|
Preparation and size of specimens, choice of radiation, and the time factor |
26 |
|
Appraisal of the X-ray diffraction powder method for nar- cotics identification |
30 |
|
Acknowledgements |
31 |
|
References |
31 |
The identification of crystalline materials by X-ray diffraction methods is a routine procedure in many laboratories. Its importance in the field of narcotics, however, does not appear to have received the general recognition that it deserves. Gross and Oberst ([5] )1 have published X-ray diffraction data for fourteen substances, including hydrates, and Hubach and Jones ([9] ) have studied dl-methadon-hydrochloride, but corresponding information on a large number of other narcotics is not available, at least in the literature.
The object of the present paper is to outline such basic principles of the interaction of X-radiation and crystalline matter as are necessary for an appreciation of the X-ray diffraction method as applied particularly to the identification of narcotics in the form of crystalline powders. The data required and the means for obtaining them are discussed briefly, and the power and limitations of the method are considered. X-ray diffraction powder patterns and tabulated numerical data for eighty-three narcotics are presented in a separate paper.
Equipment and techniques peculiar to the determination of the detailed structure of single crystals are not included in the present review although data collected by these means may also be used for identification purposes. No systematic scheme of identification based on single-crystal data, and comparable to that available for powdered materials, however, has been published although there is reason to believe that one may appear in the near future. In any case, many samples of narcotics, as normally received, do not contain crystals suitable for single-crystal measurements. Furthermore, the collection of the minimum X-ray diffraction data for the identification of a substance in the form of a single crystal frequently requires more time than is necessary when the same material is in finely powdered form.
For those who may wish to obtain more information on the X-ray diffraction powder method of identification, or who may wish to explore the use of data obtained from single crystals, a few references ([2, 6, 8, 10, 15] ) are given. No attempt has been made to compile an extensive bibliography but further references may be obtained from those cited.
1Figures in parentheses throughout the text refer to the numbered publications listed at the end of this article.
X-radiation is produced whenever rapidly moving electrons impinge on matter. An X-ray tube consists essentially of an evacuated glass envelope containing two electrodes (anode, positive; cathode, negative) across which a high potential may be applied. Electrons are accelerated by the action of the electric field and are driven against the positive electrode (anode, anticathode, target) where the rapid reduction in their velocity results in the emission of X-radiation comprising all wavelengths above a minimum value which corresponds to the instantaneous stopping of an electron moving with maximum possible velocity under the applied potential. The intensity of this continuous (or "white") radiation rises rapidly from zero, at the minimum wave-length, to a maximum and then decreases more slowly for increasing wave-lengths. As the voltage across the X-ray tube is increased, the minimum wave-length alters to lower values as does that for which the intensity is a maximum. At the same time the magnitude of the intensity at all wave-lengths increases. The wave-lengths comprising the continuous radiation, therefore, are independent of the target material, while the value of the minimum wave-length and the relative intensity at each wave-length above the minimum are governed by the applied potential.
Superimposed on the continuous radiation are intense rays of definite wave-lengths characteristic of the particular metal of which the target is composed. These characteristic X-rays arise from the disturbance of certain specific electrons in the atoms of the target material. The electrons outside the nucleus of an atom are classified in groups (shells, orbits; K, L, M, etc.) according to the amounts of energy required to remove them from the atom. X-rays of definite, characteristic wave-lengths are emitted when electrons from other groups (in higher energy levels) replace those of a given shell that have received sufficient energy to escape. Thus the K βline is emitted during the transition of electrons from the M to K shell while two lines (Kα 2, Kα 1) are produced by the transition of two electrons, with slightly different energies, from L to K shell. The excitation energy to remove electrons from a given shell is supplied by the electron beam in the X-ray tube. Since this energy must have a definite minimum value depending on the particular shell and the particular kind of atom involved, the characteristic X-rays are obtained only above a minimum applied potential and they emerge with definite wavelengths, both features being dependent on the specific chemical element making up the target. Thus the K lines of the X-ray spectrum of copper are emitted by an X-ray tube only if the potential is 9 kilovolts (kV) or more, while 20 kV is required to excite the K lines of molybdenum, and the wave-lengths of the K β, Kα 1, Kα 2 X-rays from copper are not the same as those of the corresponding rays from molybdenum.
The supply of electrons in the X-ray tube usually is furnished by a tungsten wire (filament) heated to incandescence and situated at the cathode. "Sealed-off tubes, as their name implies, are permanently closed at the factory after evacuation, while "continuously pumped" or "demountable" tubes depend on the operation of a pumping system to maintain a satisfactorily low pressure during operation. The first type is generally more popular in North America while the second is frequently used in Great Britain and on the European continent. During operation of either type, tungsten distills slowly from the hot filament and condenses on the target and on the windows through which the X-rays emerge from the tube; eventually the filament breaks. The deposit on the target gives rise to X-radiation characteristic of tungsten superimposed on that of the target metal. Under certain conditions the interpretation of diffraction patterns may be complicated by this feature. The formation of a thin film of tungsten over the windows of the tube tends to decrease their transmission for X-radiation and hence reduces the intensity of the X-ray beam obtainable under a given set of operating conditions. A remountable tube, however, can be taken apart in the laboratory for cleaning, for interchanging targets of different metals, and for replacing burned-out filaments. Thus the purity of radiation can be maintained and only one X-ray tube, with spare targets of different metals and a supply of filaments, is required for all purposes. On the other hand, an X-ray-unit with sealed .tubes generally requires much less maintenance. Capital expenditure is higher because a complete tube is required for each target material and must be replaced when the filament burns out or if the tube becomes "gassy" (i.e., if the pressure in the tube rises appreciably). Nevertheless, where loss of time due to operating difficulties is important, or where skilled technical assistance is not available, the sealedtube X-ray diffraction unit generally is preferred.
In addition to the foregoing filament, or hot-cathode, types, so-called gas tubes are sometimes employed, although less frequently now than formerly. In these the supply of electrons is obtained from the impacts of positive gaseous ions on the cathode and a small pressure (0.004 to 0.001 mm. of mercury, in the case of air) is maintained in the tube during operation. Gas tubes are relatively inexpensive and can be made in any competent machine shop. They are readily demountable for cleaning and interchange of targets, but they usually require considerable technical skill for successful operation.
Depending on the wave-length of the X-radiation desired, and on the physical properties of the target metal, potentials of the order of 35 to 60 kV usually are employed in X-ray diffraction investigations.
X-radiation belongs to the same class of phenomena as the gamma rays from radio-active sources, ultra-violet rays, visible light, infra-red rays, radio and electric waves, all of which, on the basis of classical electrodynamics, are electromagnetic waves. The wave-lengths comprising the X-ray band of the spectrum lie between those of the gamma rays and those of the ultra-violet. For purposes of comparison, the characteristic (Kα) wave-lengths of certain metals frequently used as target materials in X-ray tubes are as follows: molybdenum (Mo), 0.7107 A; copper (Cu), 1.5418 A; cobalt (Co), 1.7902 A; iron (Fe), 1.9373 A; chromium (Cr), 2.2909 A, where 1 A=10-8 cm., while the wave-length of the yellow D-line of sodium, in the visible region, is about 5890 A. Although all electromagnetic waves have a corpuscular character in many of their manifestations, attention can be confined almost exclusively to their wave nature for purposes of the present discussion.
Like visible light, X-rays travel in straight lines and are differentially absorbed by matter but, due to their much shorter wave-lengths, they can penetrate many substances opaque to visible light. To these properties are due the wide-spread applications of X-radiography for the study of otherwise hidden objects and conditions by means of shadow photographs (radiographs) and fluoroscopy.
Since X-rays are not visible they must be observed indirectly by their action on photographic emulsions or on fluorescent materials. X-radiation has marked biological effects so that suitable safety precautions for operating personnel must be observed. Finally, X-rays undergo reflection, refraction and diffraction as does visible light, but modified in scale due to their shorter wave-lengths.
For centuries it has been realized that the geometrical regularity of the forms exhibited by single crystals must be due to correspondingly regular arrangements of their constituent atoms or groups of atoms. The discovery of X-radiation by W. C. Röntgen in 1895, and the pioneering work of M. von Laue in 1912, followed and expanded immediately by W. H. Bragg and W. L. Bragg, on the interaction of X-rays with crystalline matter not only verified this belief but has led to the elucidation of the detailed structure of a vast number of crystals, many of them of extreme complexity. The application of X-ray diffraction techniques to the study of crystalline powders, with which this paper is concerned, was devised in 1916 by A. W. Hull and by P. Debye and P. Scherrer, and the X-ray diffraction powder method frequently is referred to as the Hull, the Debye-Scherrer, or the Hull-Debye-Scherrer method.
A crystal is built up of a periodic three-dimensional arrangement of atoms, ions, or groups of atoms. These repeat units may be visualized as situated at the points of a three-dimensional net, called a space lattice, such as that shown in figure 1. Thus, if an observer were sta tioned at any point (say, G) in the lattice and moved along each of any three straight lines (say, GH, GE, GC) he would encounter a series of points along each line, separated by exactly the same distance and equivalent to one another in all respects. Consequently, an observer at point G of the lattice represented in figure 1 could not distinguish his point of observation from point H, or E, or C, or any other point of the lattice; his surroundings would be identical in all cases. The three basic lines GH, GE, GC, in direction and in length, therefore, define the particular lattice of figure 1. These three lines also define the edges of a box, called a unit cell, ABCDEFGH, and the complete space lattice may be constructed by the regular stacking in three dimensions of an infinite number of these unit cells. The lengths ( a, b, c) of the edges of the unit cells may be equal or unequal as may the angles (α, β, γ) that they make with one another. An examination of figure 1 will show that the points of the lattice can be joined by other sets of lines defining unit cells of different shapes from the particular one outlined. There are, however, certain rules on which the choice of lines defining the edge lengths and angles of the unit cell in a given structure is based and all crystal structures can be referred to fourteen basic lattice types.

For the present purpose, however, it is sufficient to recognize that, regardless of how the unit cell and hence the lattice is selected, all points of the lattice lie on sets of parallel planes. The planes of each set are separated by the same perpendicular distance apart, or interplanar spacing, d, which is characteristic of that set. Thus, in figure 1, ABCD and EFGH lie on two adjacent planes of a set of equidistant parallel planes, ABEF and CDGH lie on another set with a different spacing, and so on. The plane faces of well-developed crystals are simply an outward manifestation of this fact; they are parallel to sets of planes within the crystal structure and these planes usually are those having a relatively large number of atoms per unit area.
For each crystalline material either the detailed spatial arrangement of the atoms, or their distances apart, or, in most cases, both factors are virtually unique. Thus, each crystalline substance may be distinguished by its specific collection of sets of parallel planes and, consequently, by a characteristic series of d values. Although coincidence of individual spacings may occur, only very rarely are all the spacings of one substance so nearly equal to all those of another that there is any ambiguity in their differentiation. Furthermore, the chance of such complete coincidence occurring in the case of organic compounds, such as the narcotics, which have relatively large unit cells and relatively low symmetry, is extremely small. As shown later, there is a lower limit to the values of d that can be measured experimentally through the use of X-radiation of a given wave-length. The larger the maximum value of d, therefore, the greater will be the number of interplanar spacings potentially measurable. Also, in crystals of high symmetry, several sets of planes have the same spacing (e.g., those on which lie the pairs of parallel sides of the unit cell in the cubic system) while in those of lower symmetry e.g., monoclinic) there are fewer sets of planes the spacings of which are identical, thus increasing the number of different d values that may be determined. As a general rule, therefore, if a reasonable number (say, 10 or more) spacings ( d) of the sets of planes on which the constituent atoms of a given crystalline substance are situated can be measured (with an accuracy of 1 or 2 per cent or better) the characterization of that substance for identification purposes will have been achieved.
When a narrow, parallel beam of X-rays is directed through a crystal, the electrons in the atoms of which it is composed are set into forced oscillation, and this disturbance results in each atom acting as a source from which radiates a secondary set of spherical waves of the same wave-length as the incident rays. Due to the regularity in the spatial arrangement of the atoms these scattered wavelets enhance each other in certain directions, where they are in phase, and tend to annul one another in directions in which they are not in phase. The crystal, therefore, acts as a three-dimensional diffraction grating towards the incident X-radiation and a number of diffracted rays emerge from it at certain definite angles and with certain definite intensities.
The mathematical treatment of the diffraction of X-rays by the crystal lattice is simplified by considering the effect as due to reflection of the incident X-ray beam by the sets of parallel planes of characteristic spacings ( d) on which the atoms of the crystal are located. This reflection differs from the optical case of light reflected by a plane mirror because it takes place from a set of many equidistant parallel planes simultaneously and not solely at the surface. For this reason, X-ray reflection from a crystal occurs only for certain angles of incidence; reflection of light from a plane mirror takes place at any angle of incidence. The Bragg equation, n λ = 2d sin Θ (for which a simple derivation is illustrated in figure 2) expresses the relationship between the wavelength (λ), the spacing ( d), and the angle of incidence (Θ), that must be satisfied in order that X-ray reflection shall occur. The rays reflected from successive planes must be a whole number ( n = 1, 2, 3, ....) of wave-lengths apart in order that they shall be in phase and thus enhance each other (as shown in figure 2 for three successive planes of spacing ( d)). For a given set of planes a series of reflections (first, second, third, etc., order) are obtained at different angles of reflection (Θ) for a given wave-length (λ), depending on n (i.e., depending on whether the rays from successive planes are 1, 2, 3,... wave-lengths apart). It is customary, however, to consider all reflections as first order and to write the Bragg equation in the form: λ = 2( d/n) sin Θ, or simply, λ = 2d sin Θ, thus treating the nth order reflection from a set of planes of spacing d as if it were the first order reflection from a set of planes of spacing d/n. Taking the spacings in this sense, the Bragg equation contains three variables (λ, d, and Θ) so that if any two (e.g., λ and Θ) can be fixed, or measured, experimentally the third (e.g., d) can be determined.
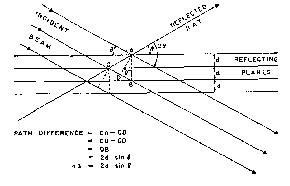
FIGURE 2
Derivation of the Bragg equation
In the Laue method a beam of X-rays comprising a continuous band of wave-lengths (the so-called "white" radiation) is directed through a stationary crystal. In this case, Θ for each set of planes in the crystal is fixed but each set reflects rays of the particular λ required to satisfy the Bragg equation for its particular value of d. More commonly, however, a beam of essentially monochromatic X-rays (fixed λ) is employed and the orientation of the crystal is varied during exposure so that different sets of planes are brought successively into reflecting position (i.e., Θ is varied continuously and the values at which reflections occur are observed).
A crystalline powder consists of a very large number of very small single crystals. If the powder is very finely divided, a small, randomly selected, sample will generally contain crystals in all possible relative orientations. Thus, or the reflection of a given wave-length λ by a particular set of planes of spacing d, at least a few individual crystals will be in such position that this set of planes makes the appropriate angle Θ with the direction of the incident beam. Therefore, each set of planes characterizing the structure of the particular substance will, in different crystals of the powder, be in correct reflecting position, and all possible reflections will occur simultaneously. Furthermore, assuming completely random orientation, individual reflections from a given set of planes, but from different individual crystals, will be distributed over the surface of a cone having the incident X-ray beam as axis, and a semivertical angle 2 Θ, governed by the interplanar spacing d and the wave-length λ. In figure 2 it will be observed that the reflected ray makes an angle 2 Θ with the incident beam. If such a set of planes be rotated about the incident beam as axis, the reflected ray will generate a cone of semivertical angle 2 Θ, as illustrated in figure 3 for a ray in the transmission region (2 Θ 1 90°) 110 and for one in the back reflection region (2Θ 2 90°). The same effect is achieved from a large number of stationary crystals in completely random orientation; at least some will be distributed in all angular positions around the incident beam while a given set of planes, in separate crystals, maintains the required angle of incidence Θ for reflection to occur. If relatively few crystals are in correct orientation, the reflections will emerge as discrete rays along the surface of the cone; if a large number of crystals are in reflecting position, the individual rays will merge to cover the surface of the cone completely and uniformly.

FIGURE 3
X-ray fiffraction comes from a crystalline powder
From the randomly orientated crystals of a powder sample, therefore, a family of coaxial cones of reflected rays is obtained. The cone angles 2Θ depend on the spacings d characteristic of the crystal structure and on the. wave-length λ of the incident beam. Now 2Θ may have any value from 0° to 180° so that observable values of Θ extend from 0° to 90° and sin Θ values cover the range from 0 to 1. Furthermore, since d = λ/2 sin Θ, the smallest measurable value of d is equal to λ/ 2. Finally, for a fixed value of d, sin Θ is directly proportional to λ so that better dispersion of the reflection cones (i.e., 2Θ values) is obtained with longer wave-lengths.
In addition to these purely geometrical features that enable a given crystalline substance to be characterized in terms of a series of interplanar spacings, each reflection has a definite intensity, determined by the particular atoms concerned, the geometrical spatial arrangement of the atoms, the wave-length of the radiation, and certain other factors. Although reflection intensities are of the greatest importance in the elucidation of crystal structures, they serve largely for verification purposes in powder identification and as a convenient basis for the classification of d values. Thus if two substances have similar structures with unit cells of almost the same size and shape, there may be some difficulty in differentiating one from the other solely on the basis of their very similar interplanar spacings d. Since their constituent atoms are not the same, however, the relative intensities of reflections from planes of similar spacings in the two substances generally will be different. A few cases have been encountered ([4] ) where two very different substances give virtually the same powder pattern both with respect to interplanar spacings and relative intensities, but such coincidences are rare.
Two general methods are employed for recording the X-ray diffraction pattern from a crystalline powder. The Geiger spectrometer utilizes the property of X-radiation of ionizing a gas through which it passes, and the powder camera makes use of the effect of X-radiation on a photographic emulsion.
The Geiger spectrometer is rapidly becoming very popular for X-ray diffraction studies. With this instrument the 2Θ values for successive reflections from a powder sample are measured in terms of the angles through which a Geiger-Müller counter must be turned about the sample as centre to receive the reflected rays. The counter consists essentially of a metal cylinder filled with a suitable gas. A potential difference is maintained between the walls and a metal wire stretched along the axis of the cylinder. When X-radiation enters the counter the gas is ionized and a discharge takes place between the wire and the walls. In this application the X-rays behave as particles and a discharge occurs for each X-photon that enters the counter. Thus the number of discharges per unit time is proportional to the rate at which X-photons are received, and, therefore, to the intensity of the X-radiation. By means of an appropriate electronic circuit the number of X-photons entering the Geiger tube is counted and the counting rates are translated into a line on a moving chart. From the distances apart of peaks on the chart, corresponding to reflections from the sample, the 2 Θ values are obtained, and after suitable calibration, intensities are determined from the heights of the peaks.
When relatively large samples are available the Geiger spectrometer is both accurate and rapid, although some skill in specimen preparation is necessary for best results. Special sample holders have been devised for specimens of a few milligrams weight. If quantities of only a milligram or less are available, as is frequently the case in a narcotics investigation, the Geiger spectrometer is less reliable and more time-consuming than the photographic method.
For very small samples, therefore, the powder camera is still the best means for obtaining a record of the diffraction pattern of a crystalline powder for identification purposes. Photographic films or plates recommended for X-ray diffraction work should be used and they should be processed with the developer and fixer specified for the emulsion selected. Over a considerable range the photographic density is directly proportional to the intensity of the X-radiation falling on the surface of the film per unit area and to the time for which the film is exposed.
The simplest powder camera consists of a pin-hole collimator for defining a pencil of virtually parallel monochromatic X-rays of about 0.5 mm. diameter, and a cassette to take a flat photographic plate with its surface normal to the X-ray beam. The diffraction cones from a powder sample intersect the flat plate in concentric circles from the radii of which, and the distance from the sample to the plate, the corresponding values of 2Θ are obtained as illustrated in figure 4. Interplanar spacings ( d) may then be calculated with the aid of the Bragg equation and a knowledge of the wave-length (λ) of the X-radiation employed. The flat plate camera, however, is seldom used. In the first place only a limited number of diffraction cones in the transmission (small 2 Θ) region can be recorded. Secondly, the length of path through which a reflected ray must travel to reach the plate increases as 2 Θ increases, thus reducing the intensity by increasing air scatter and aggravating the normally rapid decline in intensity characteristic of organic compounds. Finally, as 2 Θ increases, the reflected rays traverse the emulsion with increasing obliquity thus broadening the lines of the diffraction pattern and impairing their resolution.
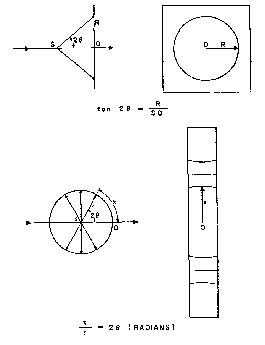
FIGURE 4
Measurement of flat plate and cylindrical film
Generally, therefore, the camera is in the form of a cylinder with the sample, preferably also of cylindrical shape, at the midpoint of the axis which is there intersected at a right angle by the collimated X-ray beam. All reflected rays lying on a circle, normal to the axis of the cylinder and passing through the incident X-ray beam, travel outwards from the specimen along radii of the camera. All such rays, therefore, traverse the same distance and pass through the cylindrical strip of photographic film, lining the inside surface of the camera, normal to its surface. With this type of camera, all possible reflections from Θ = 90° to Θ = 0° can be recorded, except for a small region at 2 Θ = 180° masked by the collimator, and a similar one at 2 Θ = 0° covered by some device for the removal of the undiffracted portion of the direct beam.
The choice of the radius of a cylindrical camera is largely one of compromise between exposure time and resolution. For a given X-ray wave-length, the larger the camera radius the greater will be the separation of the cones of reflection when they intersect the film. On the other hand, the length of path between specimen and film increases, resulting in increased scattering of the reflected rays by air molecules. This reduces the intensity so that a longer exposure is required to obtain a pattern of reasonable photographic density, and the scattered radiation appears on the film as an increase in the general background fogging.
In Great Britain a camera diameter of 90 mm. for general use, and 190 mm. for precision measurements, has been recommended by the X-Ray Analysis Group of the British Institute of Physics. A popular diameter in France is 76.4 mm. in which a length of 1 mm. on the film represents a 2 Θ angle of 1.5°. Although cameras of several diameters are common in North America, there is a trend towards the standardization of diameters of 57.3 mm. and 114.6 mm., the first for patterns in which resolution is not critical or when relatively short exposure times are desirable, the second for more complex patterns. The obvious advantage of these diameters is that 1 mm. on the film represents a 2Θ value of 2° in the first case and 1° in the second, thus reducing the conversion of film measurements into reflection angles (Θ) to simple mental arithmetic.
In the processed photograph the intersections of the reflection cones with the circular strip of film appear as curves, concave towards the point corresponding to 2Θ = 0 if 2Θ < 90°, a straight line at 2Θ = 90°, and concave towards the point corresponding to 2Θ = 180° if 2Θ > 90°, as shown in figure 4, where the determination of 2Θ from linear measurements of the flattened-out film and the known camera radius is illustrated. In actual practice, measurements (2 x) usually are made from arc to arc across the centre (0, figure 4). For cameras of 57.3 mm. and 114.6 mm., values of 2 x are merely divided by 8 and 4, respectively, to obtain Θ directly in degrees. In other cameras a less simple conversion factor must be used, or a chart may be prepared for conversion of film measurements (in mm.) to Θ (in degrees). Interplanar spacings ( d) can be calculated from the Bragg equation, or may be obtained from tables of d vs. Θ 16, or from transparent scales of d values prepared for the particular camera and wave-length used and superposed directly on the film. This last method is the most rapid since no film measurements or determinations of Θ are involved.
Several methods of mounting the film in a cylindrical camera are shown diagramatically in figure 5. In what may be termed the normal method, the ends of the strip of film are at the collimator. The undiffracted portion of the beam is prevented from fogging the film directly in front of the collimator by a beam trap of lead that absorbs the radiation, or by providing an exit tube that passes through a hole punched in the film. With the van Arkel mounting the film is reversed; the collimator goes through a hole in the film the ends of which are at the exit tube. More convenient and precise determinations of interplanar spacings in the back reflection region are possible with this type of film mounting but it is unsuitable for organic substances for which lines of large 2Θ angles usually are not observed.
The most convenient mounting is that due to Straumanis in which the ends of the film meet at a line corresponding to 2Θ = 90° and holes are punched in the film for insertion of both the collimator and the exit port. Since the distance between the holes in the unprocessed film substends an angle at 180°, its measured length after the film is developed may be used to determine the extent of any shrinkage that has occurred during processing of the film. If significant shrinkage is found, a correction is applied to the measurements of the diffraction arcs. If arcs are present in both the transmission and back reflection regions they are used to determine the effective centre of each hole. If there are none in the back reflection region, which usually is the case with narcotics, film shrinkage can be estimated by measuring across the diameter of each hole to determine the centre-to-centre distance between them and comparing this value with that obtained from similar measurements on a strip of film that has been cut and punched but has not been processed. Alternatively, the Wilson modification of the Straumanis film mounting may be employed in which the ends of the film meet at a line corresponding to a 2Θ value of approximately 40°. This ensures that measurable lines of relatively low 2Θ will bracket each hole in the film as shown in figure 5. Either the Straumanis or the Wilson method of film mounting is highly satisfactory for powder photographs of organic compounds.
For most practical purposes it is sufficient to grade the intensities of the reflections visually, in terms of the relative density of the lines of the powder pattern, as strong (s.), medium (m.), weak (w.), interpolating with v. (very), s., s.m., m.w., v.w., etc. It has, however, become customary to place these estimates on a numerical scale relative to 10, or 100, for the strongest line, thus (1), v.v.s, 100; v.s., 90; s., 80; m., 70, 60, 50; w., 40, 30; faint, 20; very faint, 10. Such relative intensities are tabulated under the symbol, I/I 1 (i.e., the ratio of the intensity of each line to that of the most intense which is referred to as the strongest, or first, line).
Visual estimations theoretically can be improved by comparison of each line of the powder photograph with a calibrated intensity scale ([7] ). Such a scale can be prepared by exposing a strip of film for predetermined times for an X-ray beam of constant intensity defined by slits to produce lines of approximately the same width as those of the powder pattern.
The use of a microphotometer, such as those employed for spectroscopic plates, is not necessary in most powder identification work. It is more time-consuming than visual estimations and may actually be misleading due to the relatively high background intensity of many X-ray diffraction powder films.
The identification of a substance from its X-ray diffraction powder data requires the comparison of its pattern with those of authenticated standards until a perfect match is found. Such a search may be carried out with the films themselves, in which case the standard powder photographs with which they are compared should be those taken in a camera of the same radius and with the same X-ray wave-length. Alternatively, tabulated data for d (independent of camera radius and X-ray wave-length) and I/I 1 may be employed.
Several years ago, Hanawalt, Rinn, and Frevel (7) proposed what has now become a standard system of identification based on values of d and I/I 1. In the A.S.T.M. index of X-ray powder data ([1] ), X-ray powder patterns are classified according to the spacings ( d) of the three lines of strongest relative intensity ( I/I 1). Separate cards for each pattern are filed in groups of limited ranges of d for their strongest line ( I/I 1 = 100). Within these Hanawalt groups they are placed in numerical order of d for the second strongest line and, where these happen to be equal, the order of the third strongest line is followed. The cards also carry the d and I/I 1 values for the complete pattern and other pertinent data about the substance. A search of the file involves selecting the Hanawalt group within which the d of the strongest line of the unknown pattern occurs, going through these cards for the d of the second strongest line and checking by means of the d of the third strongest line. If a card for a substance with the same (or nearly the same) d values as those of the unknown for the first, second, and third lines is found, the complete set of d and I/I 1 values on the card is compared with that of the unknown. In such a search and comparison some allowance, of course, must be made for permissible small variations in d due to unavoidable differences in film measurements, and in I/I 1 due to the fact that the intensity of diffraction from the same set of planes of the same substance is not independent of the wave-length but is affected to some extent by both λ and sin Θ. Furthermore, since personal differences in appraisal of I/I 1 for lines that are of almost equal photographic density may place the three strongest lines in different order, it is convenient to use a three-card set in which triplicate cards are filed in the order 1, 2, 3; 2, 1, 3; 3, 1, 2. Complete coverage by including cards for the remaining combinations 1, 3, 2; 2, 3, 1; 3, 2, 1 would make the system too unwieldy in view of the fact that the file already contains cards for about 4,800 patterns (14,400 cards in a three-card set). The data cards are available in three forms; plain for manual sorting, punched for mechanical (Keysort, Copeland-Chatterton) sorting ([12] ), and punched for machine (International Business Machines (I.B.M.), Hollerith) sorting ([11] ).
Due to the fact that the A.S.T.M. index includes data from a large number of laboratories obtained under various conditions and with different degrees of care and accuracy, it is not surprising that it should contain some erroneous entries. For this reason, and for the satisfaction of complete certainty in an identification, it is desirable to build up a standard file of diffraction patterns to be used in conjunction with it. Furthermore, in many cases there is sufficient preliminary evidence or suspicion of the identity of an unknown to warrant rapid direct comparison of the powder photograph with those of authenticated standards selected from such a file. Even when an identification is made in the normal way with the index it is a matter of convincing satisfaction to be in a position to match the photograph obtained from the unknown with one from a standard file.
For a laboratory dealing largely with organic substances it must be admitted that the present A.S.T.M. index contains a preponderance of data on inorganic materials, although the situation is improving rapidly as more X-ray diffraction powder patterns for organic compounds become available. Although it is highly desirable that any laboratory devoted extensively to identification by the X-ray diffraction powder method should have at least a one-card set of the A.S.T.M. index, a specialized narcotics laboratory at the present time probably could set up its own filing system of data cards and standard X-ray diffraction patterns of the substances most likely to be encountered that would be less costly and almost as useful.
An ideal powder pattern consists of lines that are narrow, sharply defined, and well resolved. Each line should be continuous and of uniform intensity. Background fogging should be negligible. Photographs satisfying all these criteria require special precautions that increase the time required to prepare the sample and record the diffraction pattern. In cases where identification is the primary object, the time factor usually is important and some compromise with ideal conditions must be tolerated.
Occasionally the nature of the specimen itself, such as distortion of the crystal lattice, excessive thermal vibration of the constituent atoms, or excessively small particle size, may cause diffuse and broadened lines. Improvement can sometimes be effected by annealing the specimen, by reducing the temperature during exposure in the camera, or by recrystallization, but even these expedients are not always practical or successful.
In general, however, satisfactory photographs can be obtained in a cylindrical camera with a reasonably fine ( circa 0.5 mm.) slit or pin-hole collimating system and a specimen of approximately cylindrical shape of diameter slightly less than that of the collimated X-ray beam. The specimen should be mounted with its axis coinciding with that of the camera. It should be rotated about this axis continuously during exposure to ensure that complete randomness of orientation of the crystals is achieved.
The best powder photographs are obtained from specimens in which the particle size is of the order 10-3 to 10-6 cm. Above this range the lines become spotty in appearance even with rotation of the sample because there are not sufficient crystals in a small volume to ensure completely random orientation. With particles of sizes less than a few hundred angstrom units not enough parallel planes remain in a given set, of spacing d, for the rigid validity of Bragg's equation and a sharp diffraction angle Θ diffraction takes place over a wider angular range and the lines on the photograph become broader and more diffuse.
Many samples of narcotics consist of crystals that are larger than the optimum size and these require grinding before the specimen is prepared for a powder photograph. Like many organic crystals they tend to be soft and if ground in the usual way with an agate or mullite pestle and mortar they may smear over the surface. Very small samples can be ground with less difficulty and waste by using a well-type microscope slide as mortar and a short length of drawn-down glass rod or tubing, rounded at the end, as pestle and observing the operation under a steroscopic microscope, or a fine needle may be used to break down individual crystals to a suitable size. Very fine needles may be prepared by fixing a length of tungsten wire (1 mm. diameter) in a brass handle and dripping the end of the wire into molten sodium nitrite. The taper can be regulated by the rate at which the wire is dipped into and withdrawn from the melt and it is essential to have the sodium nitrite very hot.
The speciments for the camera may be made by coating a hair or a very fine glass fibre with the powder mixed with an adhesive, or the glass fibre may be smeared very thinly with vaseline and the powder taken up on it. A support can be eliminated if the powder, mixed with a binder, is extruded by means of a piston through a small orifice in a cylinder, or the mixture can be rolled out between two microscope slides or with the fingers. Very small samples (of the order of 0.01 mg.) can be picked up in a drop of household cement on the end of a glass fibre. A form of mount that normally requires no binder or adhesive is wedge-shaped, with a small section of the edge of the wedge cut out and packed with the powder; the X-ray beam is directed through the sharp edge of the specimen. Thin-walled pyrex or Lindemann glass capillaries, or collodion tubes, may be used and are especially useful if the sample is deliquescent or efflorescent.
Care must be exercised in the selection of adhesives or binders because they may add diffraction lines of their own to the pattern. This is particularly true of vaseline if more than a trace is used. Even if truly amorphous, adhesives and binders should be employed sparingly because their presence increases the general background density of the photograph due to scattering of the radiation, and they may also produce broad diffuse haloes in the small 2 Θ region. In any case it is advisable to test a selected material by an X-ray diffraction photograph before adopting it.
The choice of a suitable X-ray wave-length depends on a number of factors. The unit cells of organic crystals usually are large so that they commonly have relatively high maximum values of d. Thus the powder pattern extends to relatively small values of 2Θ and, with very short wave-lengths, the innermost (closest to 2Θ =0°) lines may occur within the area of the film occupied by the beam trap, or exit port, of the camera, in which case they will not be recorded on the film. Furthermore, organic crystals frequently belong to the crystal classes of lower symmetry so that their patterns are characterized by a large number of lines, many of which correspond to very similar d values. Finally, the relative intensities of reflections from organic crystals tend to decrease very rapidly with increasing 2Θ angles so that the observable lines of the powder pattern generally occur only in the transmission (small 2Θ) region of the film. For these reasons it is desirable to increase the 2Θ angles as much as possible. For a given set of d values this can be done by using X-radiation of maximum wave-length since, from the Bragg equation, sin Θ is directly proportional to λ. Unfortunately, the longer wave-lengths are highly absorbed and scattered by the air in the camera. This not only increases the exposure time for a satisfactory photograph but also increases the background density of the developed film, thus rendering it more difficult to recognize and measure lines due to very weak reflections. The effect can be minimized by partial evacuation of the camera, or by filling it with hydrogen, but such procedures normally are not desirable in routine identification work. Another method of increasing the dispersion of the lines on the powder photograph is to increase the diameter of the cylindrical camera. This is of limited practical effectiveness, however, due to increased exposure times and background fogging resulting from the increased absorption and scatter of the reflected rays before they reach the film.
While many data in the A.S.T.M. index have been obtained with Mo radiation (λ (Kα) =0.7107 A), the one most commonly employed for inorganic compounds is that of Cu (λ (Kα) =1.5418 A) except in the case of substances containing a high proportion of cobalt, iron, manganese or chromium. These elements absorb much of the energy of copper radiation and radiate their own characteristic wave-lengths. This fluorescent radiation appears on the powder film as a marked increase in the background fogging. Hence cobalt (λ (Kα) = 1.7902 A), iron (λ (Kα) = 1.9373 A), and chromium (λ (Kα) = 2.2909 A) radiations, respectively, frequently are used for samples containing these elements. Unfortunately, X-ray tubes with targets of these metals usually cannot be operated at as high potentials and tube currents as those with anticathodes of molybdenum or copper. Chromium radiation frequently is employed for powder photographs of organic substances (13) because of the much improved dispersion of the lines obtained with its use. Due to air absorption and scatter, however, exposure times are increased appreciably. Thus, whereas 99 per cent of Mo Kα radiation is transmitted by 10 cm. of air, only 68 per cent of Cr Kα radiation penetrates the same distance.
FIGURE 5
EFFECT OF λ ON X-RAY DIFFRACTION POWDER PATTERNS
(Papaverine; 114.6 mm. dia. camera; Straumanis film mounting; transmission end of films; 8-hour exposures)

To ensure a clean film background, free from fog, strictly monochromatic radiation should be used but the special technique required rarely is justifiable in simple identification problems. X-ray tubes for powder investigations usually are operated at voltages that excite the characteristic K lines of the target metal with maximum intensity relative to that of the unwanted continuous radiation. The K series consists essentially of three lines, a pair (Kα 2, Kα 1) having wave-lengths separated by only about 0.004 A, and one (K β) of appreciably shorter wave-length; their relative intensities are Kα 2: Kα 1: K β= 50 : 100: 22. In practice it is very difficult to separate Kα 2 and Kα 1 although in a good powder photograph they are resolved for large 2 Θ angles as represented by the double lines in figure 5. The K β line can be removed effectively from the incident beam by interposing a suitable filter between the X-ray tube and the camera. Thus zirconium, nickel, iron, manganese, and vanadium are employed to reduce the intensity of the Kβ line of molybdenum, copper, cobalt, iron and chromium, respectively. The mass per square centimetre and the thickness of the filter are chosen so as to reduce the Kβ radiation to about 1/600 of that of the Kα this ensures that diffraction lines due to the former will not be recorded on the photographic film in the time required to obtain a good photograph of those due to the Kα radiation.
The influence of X-ray wave-length on the appearance of the powder pattern is illustrated in figure 5 by X-ray diffraction powder photographs of papaverine taken with molybdenum, copper, cobalt, iron, and chromium radiation (Kα), respectively. The same (114.6 mm. diameter) camera, the same (8 hours) exposure time and (except for the first) the same sample were used in each case. Straumanis film mounting (see figure 6) was employed but only the transmission (small 2Θ) end of the film strips is reproduced. The unsuitability of molybdenum radiation for powder photographs of complex organic substances is so obvious from figure 5 that no comment is necessary. The photographic density of the lines in the pattern obtained with copper radiation indicates that a much shorter exposure time would have been sufficient to produce a record suitable for identification; resolution is adequate in the case of the papaverine pattern but it is apparent that serious overlapping of lines would occur in those with smaller line separations. The dispersion of the lines improves progressively with the use of cobalt, iron, and chromium radiation but (for a fixed exposure time) the photographic density decreases in the same order. In this laboratory, cobalt (Kα) radiation is favoured for powder photographs of most organic substances; dispersion of the lines is better than that obtained with copper radiation, exposure times for acceptable photographs are lower than those required with iron or with chromium, and resolution usually is sufficient for identification purposes.
A relatively large sample (of the order of 0.15 mg.) was employed for the photographs reproduced in figure 5. It was mounted by taking it up in a drop of household cement on the end of a glass fibre. With cobalt radiation a pattern of sufficient density for identification can be obtained in about 5 hours from such a sample of most narcotics. An effective reduction in this exposure time by a factor of about 4 can be achieved by intensification of the X-ray film after normal processing. The necessary solutions ([3] ) are easy to prepare and the procedure adds only 30 minutes to the normal time for development and fixation.
A somewhat larger sample is required if it is necessary to place it in a thin-walled capillary tube. This method of specimen mounting, however, ensures a cylindrical shape, and usually improves the resolution of the lines on the powder photographs. Owing to absorption by the tube, exposure times tend to be increased to about 7 or 8 hours for satisfactory results. As a practical example, the powder patterns of 40 different narcotics samples have recently been obtained and compared with standard patterns in this laboratory in a period of 10 days. Two powder cameras were used with cobalt radiation, and the samples were mounted in capillary tubes. Exposures times were about 7 hours during the day and 15 hours overnight. The shorter exposures were satisfactory; the longer ones gave more complete patterns but were not strictly necessary. The films were not intensified.
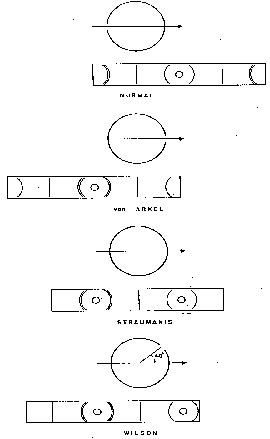
FIGURE 6
Types of film mounting
Samples are small as 0.01 mg. can be picked up in a drop of household cement on a glass fibre and their powder patterns may be obtained without recourse to special microtechniques. Exposure times, of course, are longer than those required for average specimens of the order of 0.1 mg.
Although the time required to identify the unknown sample depends on a number of factors, including the size of specimen available, it is possible in most cases to complete an identification during the afternoon if the unknown is received by 9 a.m. Alternatively, since X-ray diffraction equipment normally is provided with timers and automatic cutout devices, exposures may be started at the end of the day and the films may be processed and the patterns identified when the laboratory opens on the following morning. This compares very favourably with other methods of complete identification. The assumption is made, of course, that standard data, or standard patterns, matching those of the unknown are present in the laboratory file. If this proves to be false, identification becomes a problem of obtaining patterns from authenticated substances until one corresponding to that from the unknown is found. In such cases data from other sources and other techniques can be very helpful in indicating possible substances that should be included in the search.
The principal limitations of the X-ray diffraction powder method of identification arise from the facts that ( a) it is readily applicable only to crystalline materials; ( b) there are occasional instances of possible ambiguity due to very close similarity of patterns on account of structural similarities, solid solution, or isomorphism ([4] ); ( c) the sensitivity toward small quantities of one constituent in the presence of a large quantity of another is not very high.
Amorphous materials give rise to broad haloes and much general scatter so that identification usually is not practical, although it is possible in favourable cases. The presence of amorphous material in an otherwise crystalline sample may be inferred from the general appearance of the film but, usually, a definite statement cannot be made unless the amorphous constituent comprises the main bulk of the sample.
Almost identical patterns may be obtained from crystals which have very similar structures and in which the atoms have similar scattering effects on X-rays. This is particularly apt to be the case if the two structures are actually isomorphous. Such ambiguities, however, may be expected to be confined to the simpler inorganic structures of high symmetry (e.g., cubic, tetragonal), although some difficulty may be encountered in differentiating among isomorphous hydrohalide salts of organic bases having large molecules.
Solid solutions involve the replacement of one atom by another of comparable size in the crystal lattice. The result is a small change in the interplanar spacings ( d) that frequently can be detected only by the use of high precision techniques. This difficulty, however, usually is of major importance only in the study of metals, alloys, and minerals.
All crystalline constituents of a mixture contribute their patterns to the powder photograph so that the analysis of such a system becomes a problem of separating and identifying the individual sets of lines characteristic of each. With complex powder patterns, such as those of the narcotics, there is a high probability of coincidence of some lines, and resolution is likely to be impaired due to the lines of one constituent appearing among the already crowded lines of another. In particular cases it may be necessary to sacrifice exposure time in favour of greater dispersion of the pattern by the use of chromium radiation. When strong lines of each constituent are resolved, however, it is possible to make a semi-quantitative estimate of their relative proportions from a series of powder photographs of synthetic mixtures of known composition, although the Geiger spectrometer is more convenient for this purpose, and will yield results of greater accuracy, if sufficient material is available.
The X-ray diffraction powder method is not very sensitive towards minor constituents in a mixture because the relative intensity of each set of lines in the composite pattern depends not only on the relative proportion of each crystalline phase but also on the relative strength of each pattern under normal conditions. Thus, the strongest lines from a trace of an impurity that would give a strong pattern if it were the only substance present may be identifiable, Whereas those from a much larger proportion of a substance normally giving a very weak set of lines may not be observed. Depending, therefore, on the substances involved, the mass of one that can be detected in the presence of another varies from 1 per cent or less in the case of the simpler inorganic compounds to 5 or 10 per cent (or more) for complex and less symmetrical structures containing only light atoms such as carbon, nitrogen, and oxygen. Consequently, if the constituents in smaller proportion in a mixture are to be identified it may be necessary to effect at least a partial separation before obtaining the powder pattern. In narcotics seizures, for example, it is important that the diluent (usually lactose) be removed if the narcotic and possible adulterant (frequently quinine) are to be recognized. The importance of this point already has been dramatically illustrated in the literature (14). On the other hand, the insensitivity of the powder method to small amounts of impurities is an obvious advantage (compared, for example, with a melting point determination) if only the identification of the major constituent of the sample is required. As may be inferred, of course, the absence of lines characteristic of a particular substance does not prove that the substance is not present in a given sample; it may be there in too small a proportion for its pattern to appear on the film in the time required even for an overexposed photograph of the diffraction lines due to the other constituents.
In assessing the value of the X-ray diffraction powder method for the identification of narcotics it is important to recognize that it is not simply another method of carrying out a chemical analysis. The X-ray method, when successful, identifies a specific crystalline phase uniquely. It thus completely characterizes the phase with all those properties that may previously have been determined by other methods, and, therefore, much more is known immediately about the unknown than would be possible by ordinary analytical procedures without a protracted investigation. This is both a strength and a weakness of the method depending on the point of view. For example, if the chemical constitution is all that is desired, without regard to degree of hydration or polymorphism, the method may be considered as too sensitive because different polymorphic forms and different hydrates of the same compound give different powder patterns. Consequently, if an identification is not made, a particular substance cannot be eliminated as a possibility unless the patterns of all possible polymorphs and hydrates have been included in the search. The same situation exists in the case of compounds with the same empirical formula because, if the molecular structure is different, the crystal structure also is different, and even the powder patterns of stereochemical isomers are not identical. On the other hand, identical powder patterns are obtained from both the d and l forms of optical isomers. This limitation obviously becomes important in any attempt to use the X-ray diffraction powder method in studies involving the relative addiction potential of the two antipodes. Furthermore, it follows that the powder pattern of the dl racemoid must be the same as that of the d and l isomers if crystallization occurs under conditions resulting in the formation of a mixture of separate crystals of the d and l forms. If, however, the dl racemoid crystallizes as such, its powder pattern may, but probably will not, be identical with that given separately by the antipodes.
For the complete and positive identification of pure substances in the crystalline state the X-ray diffraction powder method possesses certain unique features. It is apparent from the foregoing discussion, however, that it is not always self-sufficient; it is frequently most powerful when used in conjunction with other techniques. The outstanding advantages of the method, particularly for the identification of narcotics, are that it is relatively easy to employ, it is non-destructive and can be applied to very small specimens, the time required compares favourably with other analytical procedures, and a permanent record in the form of a photograph or a spectrometer chart is obtained automatically. Commercial X-ray diffraction units, particularly those with sealed tubes, are not difficult to operate, and skill in preparing and mounting samples is acquired easily. The X-ray diffraction powder method is an important and very desirable addition to any analytical laboratory.
Grateful acknowledgement is made to Mrs. H. M. Sheppard, who prepared the powder photographs reproduced in figure 5, and to Mr. W. J. Forsyth, who made the corresponding intermediate negatives and prints for reproduction and who assisted in the preparation of the drawings
AMERICAN SOCIETY FOR TESTING MATERIALS: X-Ray Diffraction Data Cards and Index, Philadelphia, 1950; supplement, 1952.
002BUNN, C. W.: Chemical Crystallography, Clarendon Press, Oxford, 1946.
003CORNEY, G. M. AND SPLETTSTOSSER, H. R.: "Intensification of X-ray diffraction films to increase speed", Rev. Sci. Inst., 23, 98-99, 1952.
004FREVEL, L. K.: "Chemical analysis by powder diffraction", Ind. Eng. Chem., anal. ed., 16, 209-218, 1944.
005GROSS, S. T. AND OBERST, F. W.: "Microanalysis of opiates by X-ray diffraction", J. Lab. and Clinical Med., 32, 94-101, 1947.
006GUINIER, A.: X-Ray Crystallographic Technology (trans. by T. L. Tippel; ed. by K. Lonsdale), Hilger and Watts, London, 1952.
007HANAWALT, J. D., RINN, H. W. AND FREVEL, L. K.: "Chemical analysis by X-ray diffraction", Ind. Eng. Chem., anal. ed., 10, 457-512, 1938.
008HENRY, N. F. M., LIPSON, H. AND WOOSTER, W. A.: The Interpretation of X-Ray Diffraction Photographs, Macmillan, London, 1951.
009HUBACH, C. E. AND JONES, F. T.: "Methadon hydrochloride, optical properties, microchemical reactions, and X-ray diffraction .data", Anal. Chem., 22, 595-598, 1950.
010KAUFMAN, H. S. AND FANKUCHEN, I.: "X-ray diffraction", Anal. Chem., 22, 16-18, 1950; 24, 20-22, 1952.
011KUENTZEL, L. E.: Codes and Instructions for Wyandotte Punched Cards Indexing X-Ray Diffraction Powder Data, Wyandotte Chemicals, Wyandotte (Michigan), 1951.
012MATTHEWS, F. W.: "Punched Card Code for X-Ray Diffraction Powder Data", Anal. Chem., 21, 1172, 1949.
013MATTHEWS, F. W. AND MICHELL, J. H.: "Identification of organic compounds by use of chromium target X-ray diffraction powder patterns", Ind. Eng. Chem., anal. ed., 18, 662-665, 1946.
014PINKER, R. H.: "X-ray diffraction used in the identification of drugs", Annals of Western Med. and Surg., September issue, 595-599, 1952.
015ROOKSBY, H. P.: "X-ray diffraction techniques in the industrial laboratory", Reports on Progress in Physics, vol. 10, pp. 83-119, 1944-45.
016SWANSON, H. E.: "Tables for Conversion of X-Ray Diffraction Angles to Interplanar Spacing", App. Math., Ser. 10, National Bureau of Standards, Washington, 1950.

