The concept of conformational analysis has provided the organic chemist with a powerful tool in the investigation of stereochemical problems. This concept has been introduced by Pitzer (1) 1 and by Hassel (2) and has been popularized by Barton, who admirably reviewed the subject primarily with regard to steroid and triterpenoid chemistry (3). Several important ideas relevant to the fine stereochemistry of alicyclic systems have been contributed by Johnson (4). The value of conformational analysis, and its validity in explaining the stereochemistry of certain natural and synthetic products, are being recognized by many other workers. Thus it has been possible to employ the ideas implied by conformational analysis in a priori decisions regarding the spatial arrangements to be expected in products of various synthetic investigations, e.g., in the total synthesis of steroids (6), (7) and in the interpretation of the stereochemistry of various natural products (7), (8). Conformational analysis has been employed in alkaloid chemistry to correlate the steric relationships between the various tropane alkaloids (9), (10).
Author: David Ginsburg
Pages: 32 to 38
Creation Date: 1954/01/01
The concept of conformational analysis has provided the organic chemist with a powerful tool in the investigation of stereochemical problems. This concept has been introduced by Pitzer ([1] ) 1 and by Hassel ([2] ) and has been popularized by Barton, who admirably reviewed the subject primarily with regard to steroid and triterpenoid chemistry ([3] ). Several important ideas relevant to the fine stereochemistry of alicyclic systems have been contributed by Johnson (4). The value of conformational analysis, and its validity in explaining the stereochemistry of certain natural and synthetic products, are being recognized by many other workers. Thus it has been possible to employ the ideas implied by conformational analysis in a priori decisions regarding the spatial arrangements to be expected in products of various synthetic investigations, e.g., in the total synthesis of steroids ([6] ), ([7] ) and in the interpretation of the stereochemistry of various natural products ([7] ), ([8] ). Conformational analysis has been employed in alkaloid chemistry to correlate the steric relationships between the various tropane alkaloids ([9] ), ([10] ).
The reader who wishes to familiarize himself with the basic ideas and extensive utility of conformational analysis in cyclohexane chemistry is referred to Barton's review of the subject ([3] ). In this article certain points in the chemistry of morphine and its derivatives will be discussed in terms of the probable conformations of the substituents concerned. It will be seen that there is not sufficient data to prove unequivocally all of the conclusions reached and to extend them further to other reactions of these alkaloids. However, the general conclusions implied by this method open a fruitful field of inquiry by chemists who may be interested in supplying some of the missing evidence. To obtain part of this evidence, certain investigations are in progress in this laboratory. We beg to be forgiven for the speculative nature of a large portion of this article. These speculations will be justified if they encourage other workers to seek similar evidence in this field.
Morphine (I) consists of a tetracyclic ring system in which one of the rings (A) is aromatic, two of the carbocyclic rings (B and C) form a fused cis-octalin system and ring C and the nitrogen-containing ring D are fused in the form of a trans-octahydroisoquinoline ring system. The evidence for this steric arrangement has been reviewed in an article which recently appeared in this Bulletin ([11] ).
1Figures appearing in parentheses throughout the text refer to the numbered references listed at the end of this article.
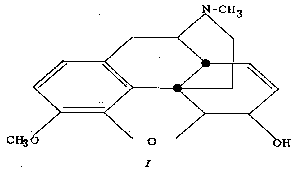
Clearly, the presence of the double bonds in the alicyclic rings, B and C, prevent these rings from attaining the true chair or boat conformations which are possible in a similar fully hydrogenated ring system. For this reason, care must be exercised in drawing the same conclusions in the cyclic system of morphine as are reached on the basis of conformational analysis of saturated systems. The same caution must be noted also in the case of compounds of the dihydromorphine series (double bond in ring C is reduced), for here also we are dealing not with a true decalin system but with a system fused to an aromatic ring through ring B. The latter, then, still shares a double bond with the aromatic ring A at their juncture, his believed, nonetheless, that the conclusions reached below are sound. With the above qualifications in mind, let us turn to the fine stereochemistry of morphine and its derivatives from the viewpoint of conformational analysis.
Neglecting, for the moment, the oxide ring of morphine, structures II and III represent the structures of tetrahydrodesoxycodeine and β-tetrahydrodesoxycodeine, respectively.
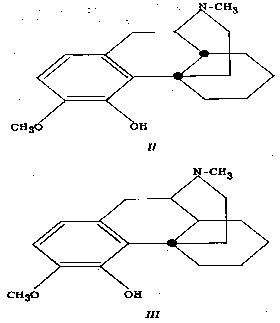
In tetrahydrodesoxycodeine (II) we have the steric arrangement present in morphine itself, i.e., rings B and C are cis-fused, while structure Ill is representative of the abnormal arrangement present in certain synthetic or degradation products, in which rings B and C are trans-fused, e.g., in - β dihydrothebainone.
OXIDATION OF ALCOHOLS IN THE MORPHINE SERIES TO KETONES
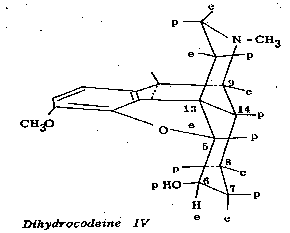
Rapoport and coworkers ([12] ) have found that certain alcohols of the dihydrocodeine series are readily oxidizable to ketones by means of the benzophenone-potassium t-butoxide system, whereas certain isomeric compounds are, for all practical purposes, not oxidized. Thus, it was found that dihydrocodeine (IV) in which the hydroxyl group at C 6 is polar, affords dihydrocodeinone in 83 per cent yield, while dihydro isocodeine (V), its C 6 epimer (equatorial hydroxyl group) gives this ketone in only 3 per cent yield.
Similarly, dihydro allopseudocodeine (VI) (polar hydroxyl at C 8) is oxidized to dihydro pseudocodeinone in 40 per cent yield but dihydro pseudocodeine (VII) (equatorial hydroxyl at C 8) is recovered unchanged under the conditions of the oxidation. Dihydromorphine (VIII) with a polar hydroxyl group at C 6, is oxidized analogously to dihydromorphinone in 71 per cent yield. These experimental results are in accord with the general experience in the preferential oxidation of polar alicyclic alcohols (cf. 3).
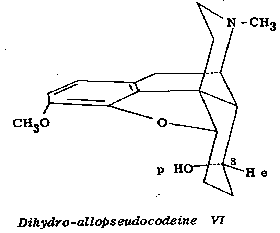
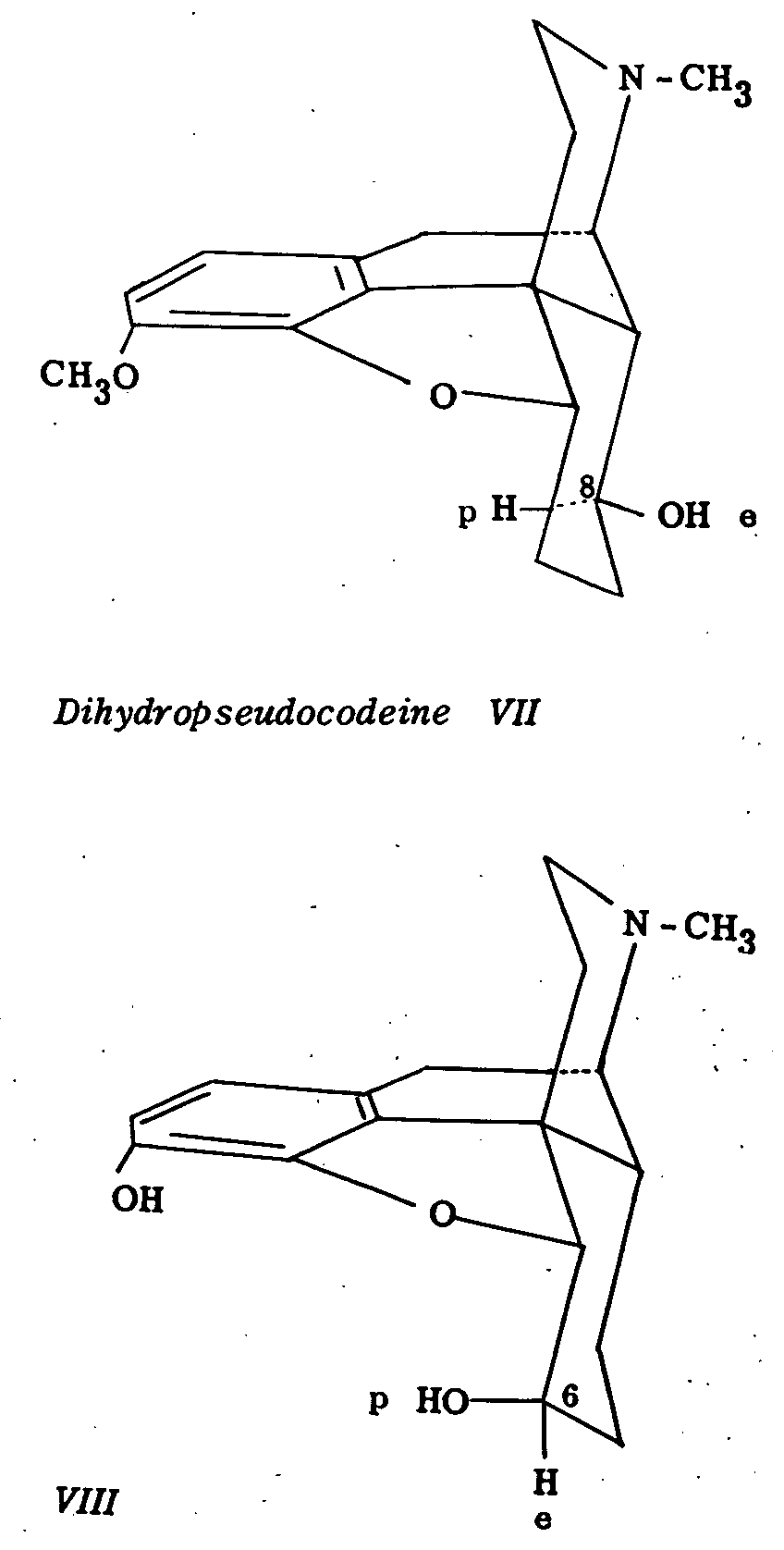
Table I summarizes the conformations of the respective substituents in various compounds of the morphine series.
|
Compound |
Substituent |
Position |
Conformation |
|
Codeine |
OH |
6 |
p |
|
Isocodeine |
OH |
6 |
e |
|
Pseudocodeine |
OH |
8 |
e |
|
Allopseudocodeine |
OH |
8 |
p |
|
Dihydrocodeine |
OH |
6 |
p |
|
Dihydro isocodeine |
OH |
6 |
e |
|
Dihydro pseudocodeine |
OH |
8 |
e |
|
Dihydro allopseudocodeine |
OH |
8 |
p |
|
α-chlorocodide |
Cl |
6 |
p |
|
β-chlorocodide |
Cl |
8 |
e |
|
Bromocodide |
Br |
8 |
e |
|
Iodocodide |
I |
8 |
e |
Since Rapoport and Payne ([13] ) have unequivocally proved the stereochemical relationship between the C 5-O ether bond and the C 6-O hydroxyl bond in codeinc to be cis- with respect to each other and since the only way one may construct the ether bridge in the cis-fused B,C ring system (see structure I), is with the C 5-O ether bond in the equatorial conformation, it necessarily follows that the C 6-hydroxyl group is polar.[2] Isocodeine, then, must be the isomer with the equatorial C 6-hydroxyl group. These facts coupled with the oxidation results for the corresponding dihydro-isomers show that the presence of the C 7-C 8 double bond in the unhydrogenated compounds does not change their molecular shape to the extent that conformational analysis fails as a tool of diagnostic importance. Direct observation of Barton models [2] leads to the same conclusion. If it may be equally assumed that a C 6-C 7 double bond in the morphine ring system will not upset the conformational applecart, then the assignments made for pseudocodeine and allopseudocodeine in table I are also correct on the basis of the oxidation data for the respective dihydro-derivatires. Definitive evidence regarding this point could easily be obtained through oxidation studies directly with pseudocodeine and with allopseudocodeine.
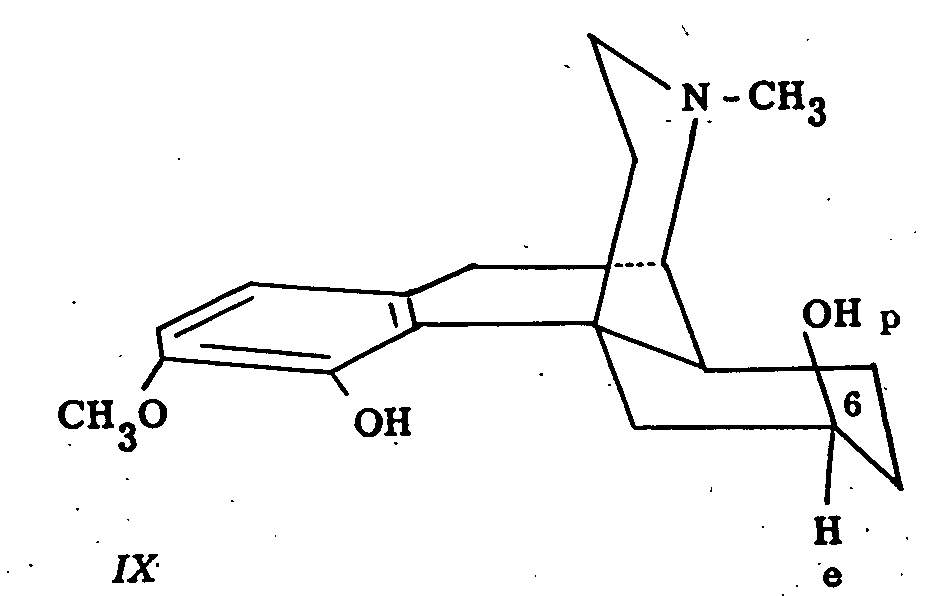
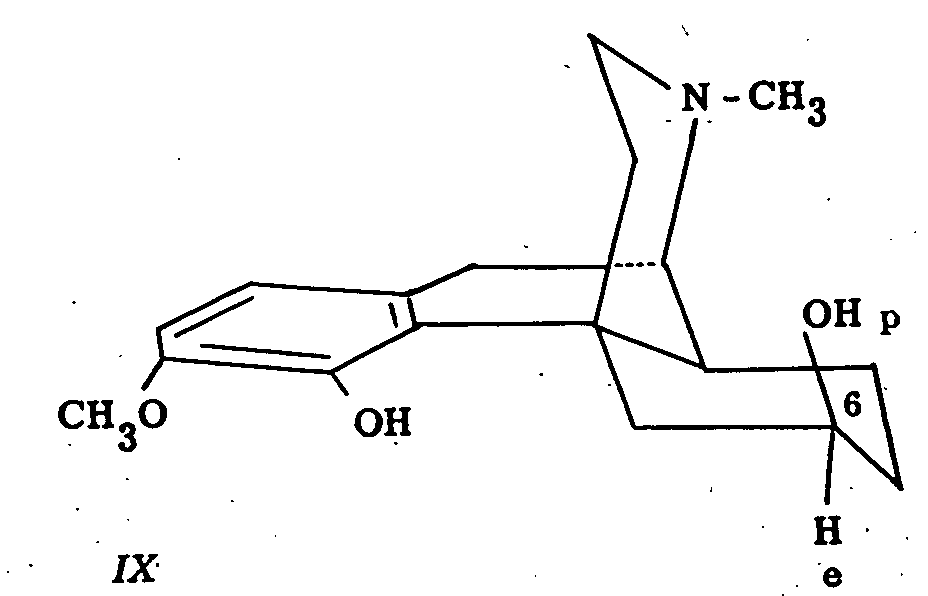
Further oxidation experiments in this field indicate that in β-dihydrothebainol (IX), the C 6-hydroxyl group is polar since this compound was oxidized to β-dihydrothebainone by means of the benzophenone-potassium t-butoxide system ([14] ).
REDUCTION OF KETONES
Sufficient data has accumulated to justify the generalization made by Barton ([3] ) that for non-hindered ketones, reduction with lithium aluminium hydride gives a greater proportion of the equatorial alcohol. Reduction with alkoxides gives more of the polar alcohol than is obtained by other methods (although this epimer is not necessarily the major product) with the exception of catalytic reduction in the presence of acid. The latter method usually affords the polar alcohol.
Dihydrothebainone (X) is reducible under a variety of conditions to the two possible epimeric dihydrothebainols ([15] , [16] , [17] ).

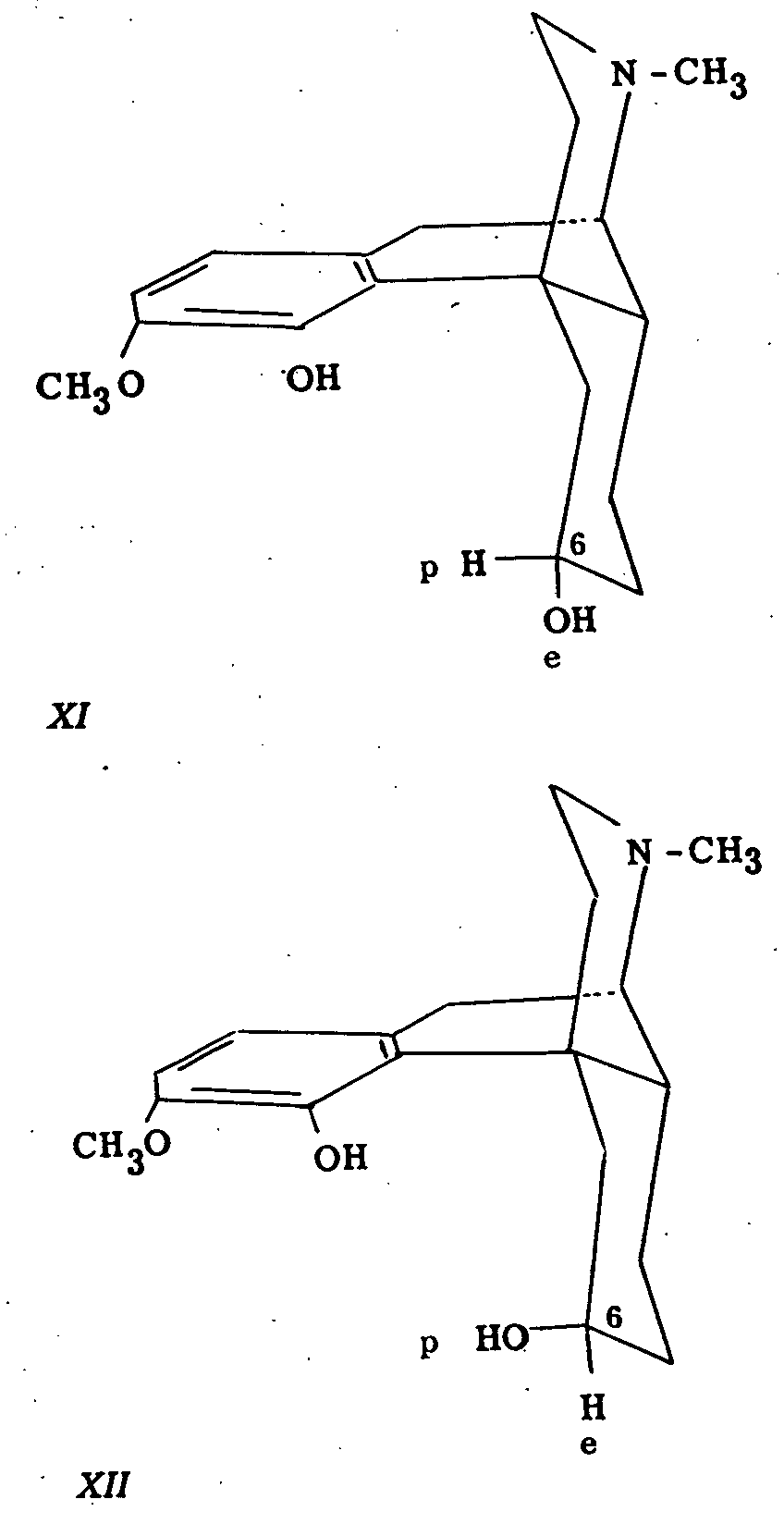
Dihydrothebainol A is obtained by electrolytic reduction of dihydrothebainone or by reduction with sodium amalgam. Dihydrothebainol B is obtained by reduction of the ketone in the presence of a platinum catalyst and hydrochloric acid. It might be argued from the following analogy to the steroids that dihydrothebainol A is the isomer in which the C 6-hydroxyl group is equatorial (XI) whereas in the B isomer the corresponding group is polar (XII).

Coprostanone (XIII) (rings A and B cis-fused) on reduction with platinum in neutral solution gives epi-coprostanol (XIV) (equatorial hydroxyl) while reduction in acid solution gives coprostanol (XV) (polar hydroxyl). Coprostanol upon treatment with sodium alkoxide at 180° is converted to epicoprostanol, indicating that in the presence of alkali the stable isomer is the one bearing the equatorial alcoholic function.
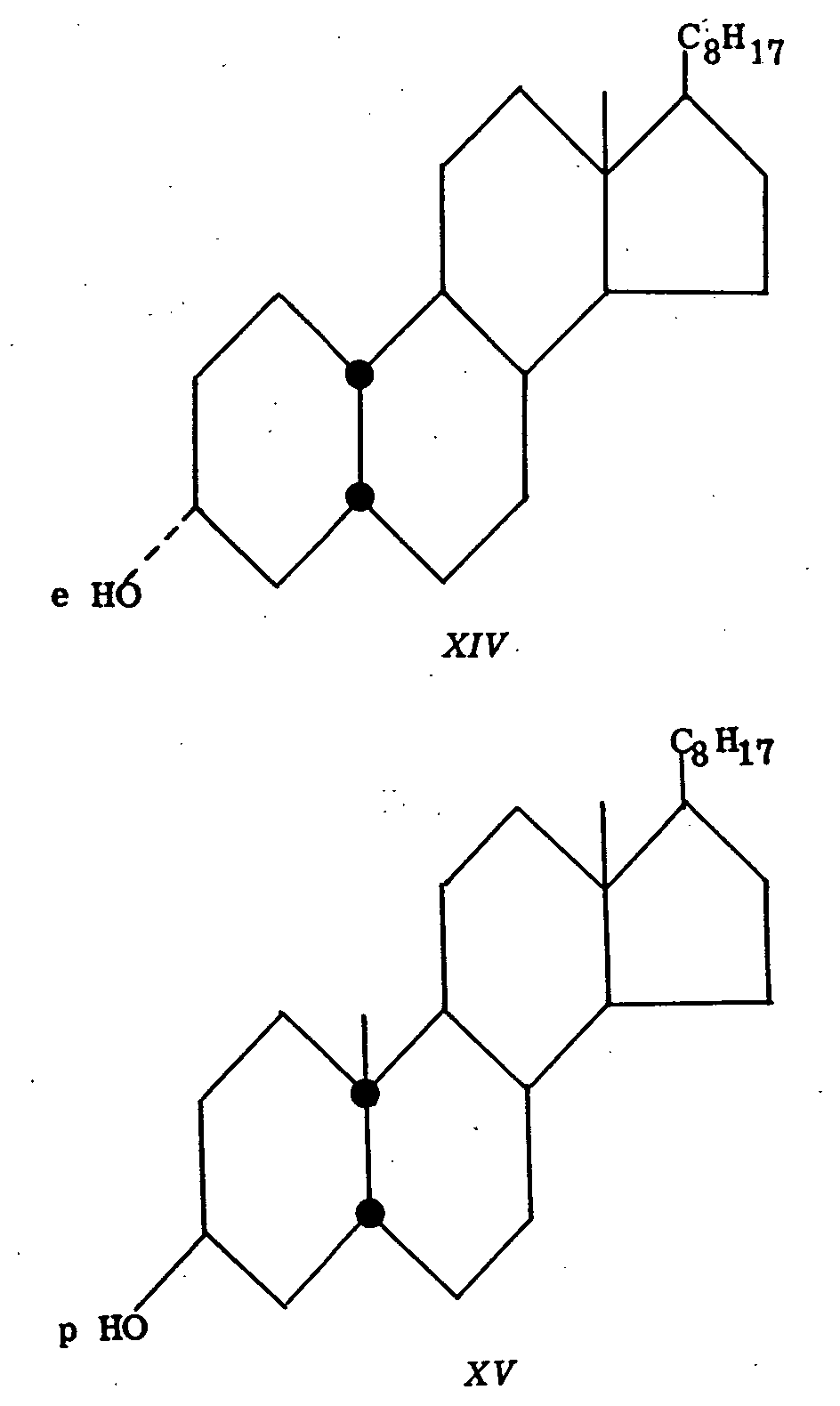
This analogy is particularly attractive since behavior of dihydrothebainone (X) with rings B and C cis-fused, upon bromination, also parallels that of coprostanone (XIII) and other steroids in which rings A and B are cis-fused. Thus, bromination of coprostanone yields 4-bromocoprostanone. Neglecting the product of bromination of dihydrothebainone with one mole of bromine, which results in the introduction of a bromine atom in the aromatic ring (at C 1, a position not germane to this discussion), the second mole of bromine effects bromination at C 5 and not at C 7. This, indeed, forms the basis for the method of cyclization of dihydrothebainone to dihydrocodeinone ([18] ).
If the above analogy to steroids is warranted, then dihydrothebainol A bears the equatorial hydroxyl group while dihydrothebainol B has a polar alcoholic function. It would therefore be expected that the B isomer would be oxidized to dihydrothebainone much more readily than the A isomer by means of the benzophenonepotassium t-butoxide system.
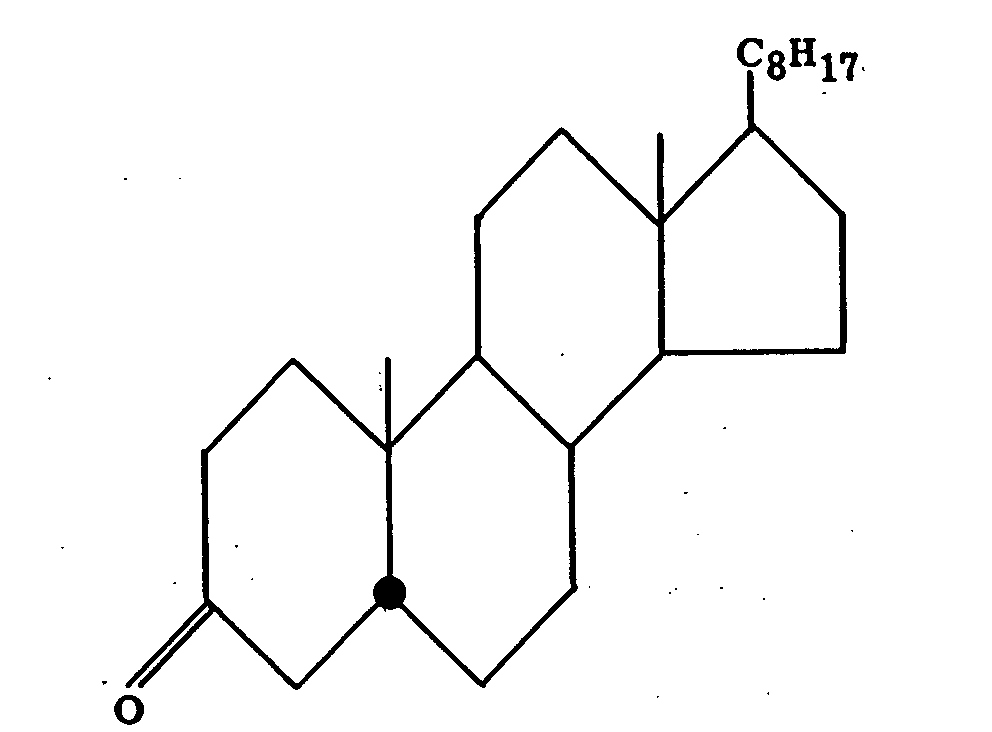
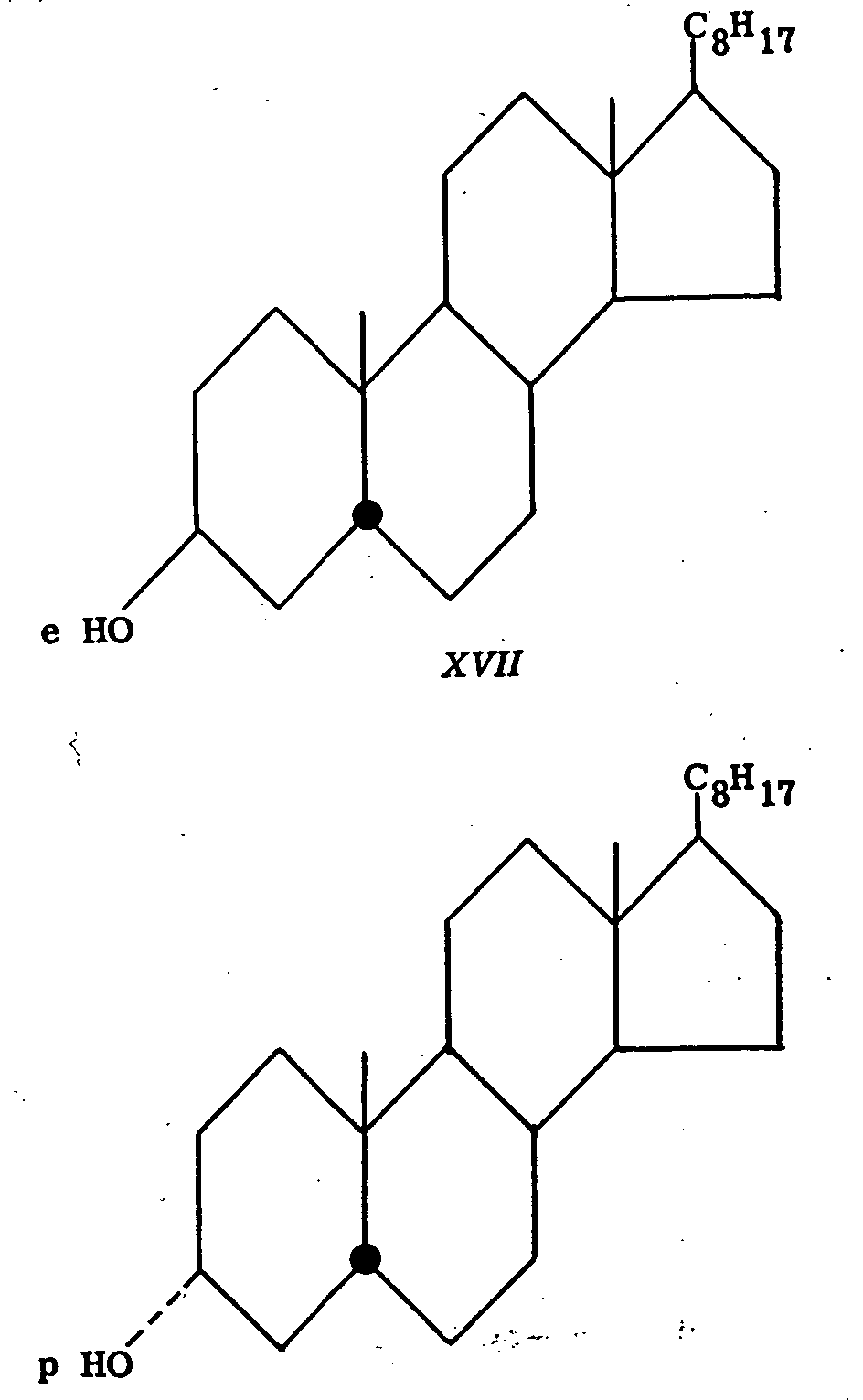
Coprostanone upon reduction with lithium aluminium hydride affords the 3α-alcohol (XIV) (equatorial hydroxyl) in 94 per cent yield and the 3 β-epimer (XV) (polar hydroxyl) in only 4 per cent yield. Similarly, cholestanone (XVI) gives 91 per cent of the 3 β-alcohol (XVII) (equatorial hydroxyl) and 4 per cent of the 3α-isomer (XVIII) (polar hydroxyl) (19).
XVI
Aluminium isopropoxide reduction, the method favoured when an optimal amount of polar alcohol is desired, of dihydrocodeinone gives, nevertheless, up to a 94 per cent yield of dihydro isocodeine (equatorial hydroxyl) ([20] ) but catalytic reduction of this ketone gives only dihydrocodeine (polar alcohol) ([21] ). Furthermore, dihydrocodeine is isomerized to dihydro isocodeine in 30-65 per cent yield by means of potassium or aluminium isopropoxide ([20] ). The reduction of codeinone by means of lithium almninium hydride has not been reported although reference has been made to the successful oxidation of codeine to codeinone by means of an alkoxide-acceptor ketone combination ([22] ). The reduction of 1-bromocodeinone with lithium aluminium hydride has been accomplished by Gates and Tschudi ([14] ) but although isocodeine might be expected to be the major product on the basis of the foregoing discussion, codeine (polar hydroxyl) was obtained. Mr. Dov Elad, in this laboratory, has reduced dihydrothebainone with both lithium aluminium hydride and sodium borohydride. In this case epimeric mixtures were obtained but the isomer, m.p. 142°, presumed to be dihydrothebainol A, predominated in the borohydride reduction while the isomer, m.p. 166-168°, presumed to be the B isomer, predominated in the lithium aluminum hydride reduction. Gates ([14a] ) has recently reported the stereospecific reduction of codeinone to codeine by means of borohydride. The reason for this result is not immediately apparent.
XVIII
Flowsheet I summarizes the relationships between codeine and its isomers and the chlorocodides. Stork ([23] ) has brilliantly interpreted the above interconversions on the basis of present knowledge of the mechanisms involved. It is therefore unnecessary to repeat the arguments here.
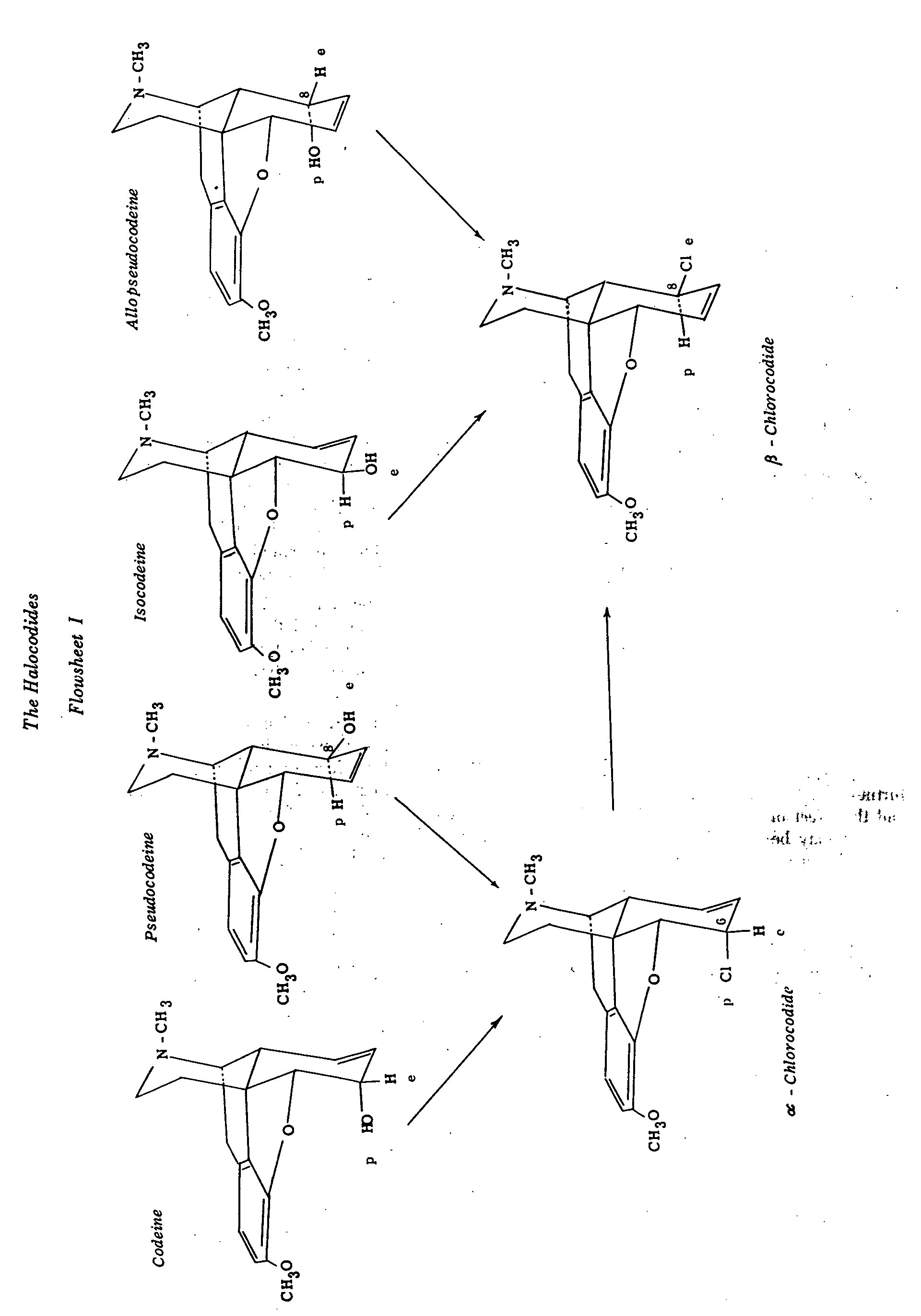
Table II summarizes the data obtained on the solvolysis of α- and β-chlorocodides in aqueous acetic acid ([24] ).
|
Solvolysis of |
Isocodeine (e-OH) |
Pseudocodeine (e-OH) |
Allopseudo codeine (pOH) |
Total equotorial products Percent |
|
α-chlorocodide |
||||
|
(p-C1) |
25 | 45 | 70 | 15 |
|
β-chlorocodide |
||||
|
(e-Cl) |
55 | 10 | 65 | 20 |
The significant conclusion from table II, from the viewpoint of conformational analysis, is that with α-chlorocodide (polar chlorine) the ma jor products are those with equatorial hydroxyl groups. With β-chlorocodide (equatorial chlorine), again the major portion of the solvolysis products consists of those with equatorial hydroxyl groups. In no case is codeine (polar hydroxyl) formed in isolable amounts; α-chlorocodide (polar chlorine) itself, is less stable than β-chlorocodide (equatorial chlorine) and is converted to the latter by means of various reagents ([25] ).
The fact that only one bromocodide and one iodocodide exist, having what is presumed to be the β-halocodide structure ([26] ), may also be explained by the fact that the halogen in this conformation is equatorial. While in α-chlorocodide, a chlorine atom is capable of existing in the polar conformation, the bromine or iodine atoms due to their greater size would not be as readily accommodated and any α-isomer, if formed, apparently prefers to rearrange allylically to the more stable equatorial conformation at C 8.
The reaction of thionyl chloride with alcohols is much more complex than had at one time been believed and may proceed by different mechanisms ([27] ). When further studies have been carried out with this reagent and the effect of reaction conditions has been better defined, it may be fruitful to repeat the work involved in the cycle summarized in flowsheet I. This would not only verify Stork's arguments in this regard ([23] ) but would also permit a stepwise test of the a priori tenets of conformational analysis.
No work has been reported on the relative acylation rates of alcohols of the morphine series. A study of the rates of esterfication of a set of alcohols such as codeine, isocodeine , pseudocodeine and allopseudocodeine and the relative rates of hydrolysis of the esters would furnish additional evidence for the conformations of the hydroxyl groups in these isomers. It would be expected that the rates of esterification would be in the order: lsocodeine>Codeine
Pseudocodeine> Allopseudocodeine
The reverse order should hold for the relative rates of hydrolysis. Similar orders of reactivities would be expected for the corresponding dihydro-derivatives.
It would also be interesting to note whether chromatography of polar and equatorial epimers in this series would give further experimental support for the generalization that polar alcohols are less absorbed than their equatorial isomers. We are aware of one case of clearcut separation of a pair of epimers which has been reported. As expected, dihydrocodeine was eluted readily from a column of alumina. Dihydro isocodeine had to be recovered from the colunm by means of dilute acid ([20] ).
Regularities in the infra-red absorption of various alcohols and their esters should be observed when data is accumulated in the morphine series, in analogy, for example, to the regularities observed in the steroids (cf. 3).
In this article certain results in morphine chemistry have been evaluated by the application of the method of conformational analysis. It is believed that the implications of this concept would lead to important confirmations of stereochemical relationships if evidence were sought on this basis in the field of morphine chemistry.
2We are grateful to Dr. D. H. R. Barton, who enabled us to obtain a set of the models which he designed. These models are very helpful in clearly illustrating the spatial relationships in alicyclic compounds.
C.W. BECKETT, K. S. PITZER AND R. SPITZER: J. Amer. Chem. Soc., 69, 2488 (1947).
002O. HASSEL AND B. OTTAR: Acta Chem. Scand., 1,929 (1947).
003D.H.R. BARTON: J. Chem. Soc., 1953, 1027.
004W.S. JOHNSON : Experientia, 8, 315 (1951); J. Amer. Chem. Soc., 75 1498 1953.
005R.B. WOODWARD et al.: J. Amer. Chem. Soc., 74, 4223 (1952).
006L. H. SARETT et al.: ibid., 74, 4974 (1952) and preceding papers in this series.
007D.H.R. BARTON AND N. J. HOLNESS: J. Chem. Soc., 1952, 78.
008D. H. R. BARTON AND E. MILLER: J. Amer. Chem. Soc., 72, 1066 (1950).
009G. FODOR AND K.NADOR: Nature, 169, 462 (1952); J. Chem. Soc., 1953, 721; G. FODOR AND O. KOVACS: ibid., 1953, 2341.
010G. FODOR: Nature, 170, 278 (1952).
011D. GINSBURG: Bulletin on Narcotics, vol. V, No. 4.
012H. RAPOPORT et al.: J. Org. Chem., 15, 1103 (1950).
013H. RAPOPORT AND G. B. PAYNE: ibid., 15, 1093 (1950).
014M. GATES AND G. TSCHUDI: J. Amer. Chem. Soc., 74, 1109 (1952).
014aM. GATES: ibid., 75, 4340 (1953 ).
015E. SPEYER AND S. SIEBERT: Ber., 54, 1519 (1921).
016A. SKITA et al.: ibid., 54, 1560 (1921).
017D. E. MORRIS AND L. F. SMALL: J. Amer. Chem. Soc., 56, 2159 (1934).
018C. SCHOEPF AND T. PFEIFER: Ann., 483, 157 (1930).
019C.W. SHOPPEE AND G. H. R. Summers: J. Chem. Soc., 1950, 687.
020M.M. BAIZER et al: J. Org. Chem., 16, 543 (1951).
021L.F. SMALL et al.: J. Amer. Chem. Soc., 58, 1458 (1936).
022Ref. 14, footnote 6.
023G. STORK: The Alkaloids, vol. 2. pp. 180-185, Academic Press, New York, 1952.
024L. KNORR AND H. HOERLEIN: Ber., 41,969 (1908).
025H. L. HOLMES: The Alkaloids, vol. 2, p. 62, Academic Press, New York, 1952.
026Ref. 23, p. 183.
027E. R. ALEXANDER: Ionic Organic Reactions, pp. 93, 279, Wiley, New York, 1950.

