Part IIIA. The Ultraviolet Spectrophotometric Method
TABLE OF CONTENTS
LIST OF TABLES
1. INTRODUCTION, STATEMENT OF OBJECTIVE
2. ORIGIN OF ABSORPTION SPECTRA
3. INSTRUMENTATION
4. THE BECKMAN SPECTROPHOTOMETER (13)
5. SPECTROPHOTOMETRIC IDENTIFICATION
6. HISTORICAL REVIEW OF ULTRAVIOLET ABSORPTION SPECTROPHOTOMETRY RELATED TO NARCOTIC ANALYSIS
7. QUANTITATIVE SPECTROPHOTOMETRY
8. THE INFLUENCE OF PH ON ULTRAVIOLET SPECTRA OF DRUGS AND ITS USE IN THEIR DETERMINATION
9. TREATMENT OF DATA
10. CONCLUSIONS
Author: Charles G. Farmilo
Pages: 18 to 41
Creation Date: 1954/01/01
|
Section |
Page | |
|---|---|---|
| List of tables | 18 | |
|
1. |
Introduction, statement of objective |
18 |
|
2. |
Origin of absorption spectra |
18 |
|
3. |
Instrumentation |
19 |
|
4. |
The Beckman spectrophotometer |
23 |
|
5. |
Spectrophotometric identification |
26 |
|
6. |
Historical review of ultraviolet absorption spectro- photometry related to narcotic analysis |
26 |
|
7. |
Quantitative spectrophotometry |
34 |
|
8. |
The influence of pH on ultraviolet spectra of drugs and its use in their determination |
36 |
|
9. |
Treatment of data |
38 |
|
10. |
Conclusions |
39 |
|
Table |
Page | |
|---|---|---|
|
I |
Combinations of filter, phototube and source in operating the Beckman spectrophotometer |
25 |
|
II. |
Csoskan's classification of opium alkaloids and spectra (43) |
32 |
|
III. |
Spectrophotometric data of opium alkaloids, according to Csoskan (43) |
32 |
|
IV. |
Buffer composition and absorbency, according to Morgan (1949) |
33 |
|
V. |
Effect of pH on absorption maxima of sulfanilamide |
37 |
|
VI. |
Absorption maxima, minima and extinction values (E 1%/1 cm) for some narcotics, according to Elvidge, 1940 |
37 |
|
VII. |
Effect of pH on the absorption maxima and minima of papaverine HCl with extinction values (E 1%/1 cm ), from Foster and MacDonald (51) |
37 |
|
VIII. |
Key points in the absorption spectra of salicyclic acid and aspirin, after Edwards 1950 |
38 |
|
IX. |
Relation of concentration to absorbency (34) |
39 |
Absorption spectrophotometry offers a means of analysis which supplements the information obtainable by colour and crystal tests and allows the rapid investigation of new classes of narcotics.
Emission spectroscopy and absorption spectroscopy are closely related and instrumentation for both is quite often found in the same laboratory. Before discussing the specific application of ultraviolet absorption spectra to narcotic analysis, one should first know of the origin of absorption spectra, become familiar with the general components of the instruments used for measuring absorption spectra and understand the laws governing the attenuation of radiant energy passing through a homogeneous, isotropic, non-metallic medium.
Emission spectra consist of sharp discrete lines arising from the transition of electrons from a term level of higher energy to a term level of lower energy in an atom or ion. These term levels can be defined by a principle quantum number, n; an azimuthal quantum number, 1; a spin quantum number, s; and the resultant angular momentum, j. The term levels of molecules are designated by a similar system of symbols where A is analogous to the azimuthal quantum number, Σ the spin quantum number, and Ω is similar to the resultant angular momentum. In emission spectroscopy the electrons are pushed or "excited" to the higher energy levels by energy supplied by means of electrical discharge. The amount of energy absorbed in this process is small and is not normally measured, since it is the energy emitted by the excited atoms which serves to qualitatively identify the atoms and to serve for their quantitative estimation. An application of emission spectrography in the narcotic analysis for the determination of countries of origin of opium samples by means of the composition of the opium ash has recently been made by Bartlet and Farmilo ([1] ).
Absorption spectroscopy is a means of investigating narcotics by the energy absorbed in raising the narcotic molecule to excited states. While atoms and ions also demonstrate a similar property of energy absorption, it is more generally utilized in the determination of molecules, or particular groups of atoms in a molecule. The molecule is made up of several atoms and its total energy is the sum of the translational energy of the entire system, the vibrational energy, the rotational energy and the electronic energy of its components. Consequently, the energy terms for molecules are more complicated than for atoms; each spectral line of the atomic emission spectrum is replaced by a system of bands for the molecular absorption spectrum. (Two examples of atomic emission spectra and molecular band spectra are given in figures 2 and 13). Energy transitions which involve change of rotational energy alone involve relatively small amounts of energy and correspond to bands in the extreme infrared or microwave regions. (The origin of these spectra are discussed in part IV A of this series.) The vibrational energies are larger, so that changes in vibrational (and rotational) energy lead to bands in the near infrared. Transitions involving electronic rearrangements in molecules may lead to absorption in any region of the spectrum. The systems used in analytical spectrophotometry absorb energy between 2,000? (200 mµ) and 10,000 ? (1,000 mµ). In general the region of the spectrum in which an absorption band occurs is determined by the electronic transition, the gross structure of the system depends on the vibrational energy changes, and the fine-line structure of the individual bands depends on the rotational energy changes.
Since qualitative spectrophotometry involves measurement of the energy absorbed by a sample as a function of the wave-length, and quantitative determinations depend upon measurements of the absorbed energy at a fixed wave-length, a spectrophotometer must consist of three essential components. These parts are a source of radiant energy, a means of dispersing the energy according to its frequency, a mechanism for detecting and estimating the amount of energy passing through the sample. In the diagram (figure 1) entitled "How a spectrophotometer works" these parts and their relation to one another can be seen from left to right in the diagram. ([3] ) See figure 1.
In the ultraviolet and visible portions of the electromagnetic spectrum, three types of sources of radiation are normally used. For the ultraviolet region from 200-400 mµ, the continuum produced by a hydrogen discharge has been widely used (Lawrence and Edlefson, 1930, and Kistiakowsky, 1931). This source gives a continuous supply of radiation below about 350 mμ, and is readily adaptable for use with instruments that contain phototubes or photomultiplier tubes as radiation detectors.
Such sources are operated at hydrogen pressures ranging from 1 to 10 millimeters of mercury, at applied voltages from 3,000 to 5,000 volts, and at a fraction of an ampere to several amperes. A hot cathode, hydrogen discharge tube has been described by Allen and Franklin, 1939, 1941, ([4] ) which operates at 80 volts and about 1.3 amperes. A ribbon filament incandescent lamp operating at a colour temperature of 2,400 to 2,800 degrees K gives a continuous source of radiation which may be used down to about 320 millimicrons. This limit is imposed by the glass envelope surrounding the filament.
A condensed spark between tungsten-steel electrodes is the most widely used source in photographic absorption spectrophotometry. This was the source employed for making all the benzene spectra illustrated in figure 2. In figure 2, the high intensity of the ultraviolet emission, the richness of the lines, and their fairly even distribution over the entire spectral range, freedom from wandering, and the localization of the luminous area in a small region may be seen. It is for these reasons that the tungsten-steel spark is preferred for work in the ultraviolet. The discontinuous character of the spectrum and the wide range of intensities between the strong and weak lines are disadvantages of this source. These two properties make the spectral image unsuitable for the detection of fine structure in absorption bands. The graph in the lower right hand corner of figure 2 was constructed from the spark-absorption spectra shown in the upper right hand corner. The graph in the lower left hand corner was constructed using a modern recording ultraviolet absorption spectrophotometer having as radiant energy source, a continuous discharge source.
Spectral dispersion and isolation in the region between 200 and 1,000 millimicrons may be obtained by means of refraction through a prism; by the diffraction and interference associated with a fine line grating, or the spectral isolation may be effected by means of optical filters, there being no dispersing system in the ordinary sense.
Glass or quartz prisms of 60 degrees refracting angle, or equivalent prisms have been widely used in prism spectrophotometers for the ultraviolet and visible regions. Recently a spectrophotometer (Beckman model B) has become available which uses a Fery prism ([3] ). Some instruments have been designed with a double prism monochromator to reduce stray light.
Gratings have been used to obtain dispersion of the radiant energy, with both transmission and reflection types being of value. A transmission grating, double monochromator spectrophotometer is claimed to have an average stray radiation of one per cent or less. However, in general, a grating monochromator has a higher relative intensity of stray radiation than a prism dispersing system. Cary and Beckman, 1941 ([5] ), described a prism dispersing instrument, which will be described in detail later, since it is the instrument on which all the spectra of narcotics described in part III B of this series were obtained.
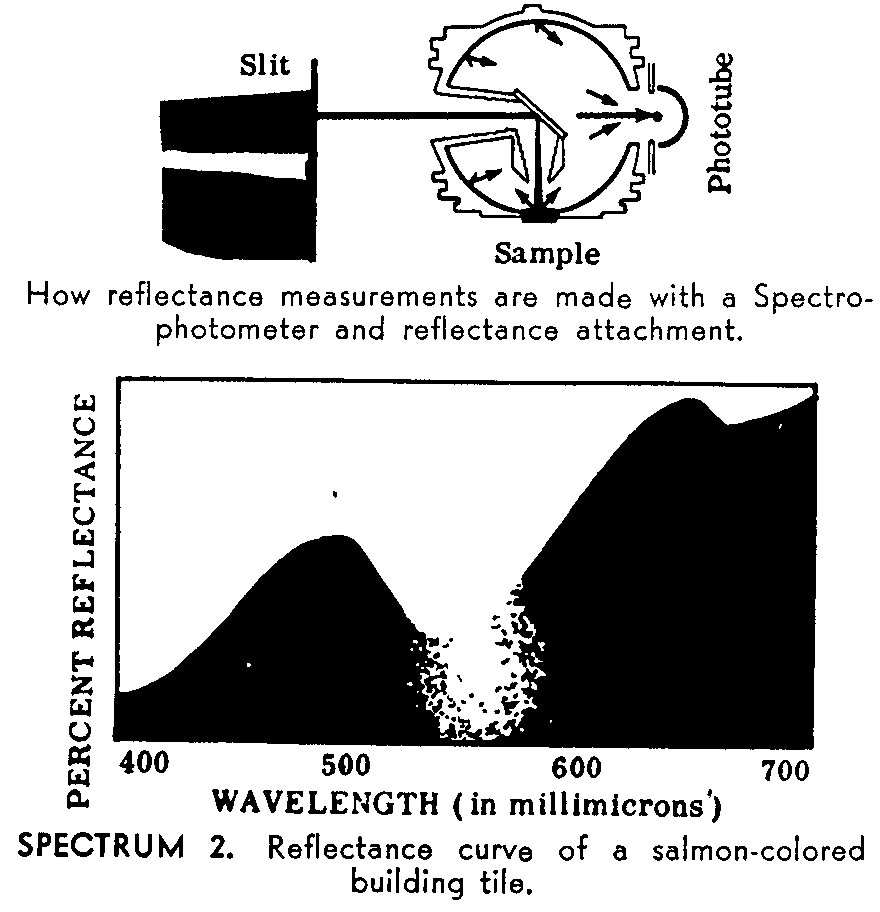
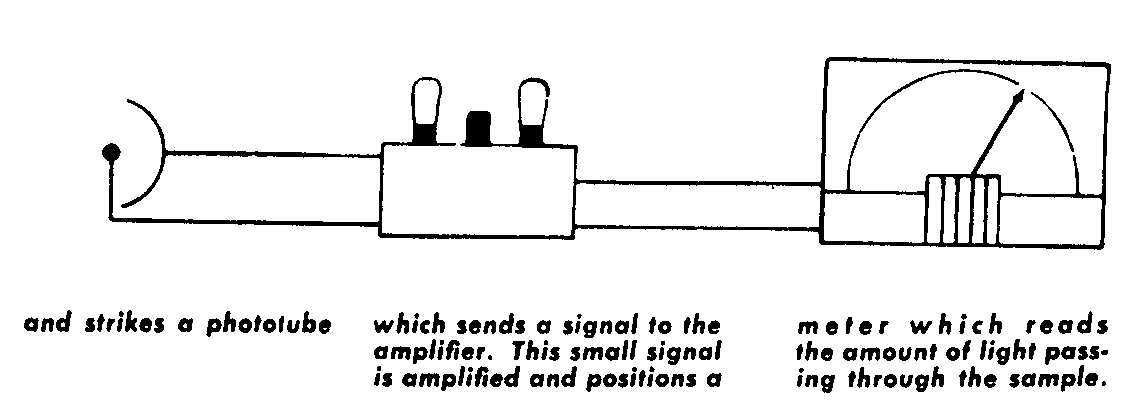
For visible light, a regular light bulb is used. Ultraviolet and infrared require special radiation sources. A large number of all measurements are made in the ultraviolet and the infrared regions of the spectrum. The desired color (wavelength) is selected by a knob which rotates the prism, causing the spectrum to move along the slit so that a different band of color falls on the slit opening and reaches the sample. The slit width is adjustable, allowing any band width of color to be chosen. The slits can be made extremely narrow to give high "resolution"--an important performance feature of a Spectrophotometer. Some of the light rays which enter the sample displace atoms from their normal position in a molecule and make them vibrate. The wavelengths which cause this vibration are absorbed by the sample. Light passing through the sample strikes the phototube where it is changed into an extremely small electric signal. The signal is then sent to an amplifier, similar to the one in an ordinary radio. By greatly increasing the strength of the minute signal from the phototube, the amplifier eliminates the need for delicate parts, permitting an instrument design which is resistant to shock and vibrations. The amplified signal positions the needle of an accurate meter to read the exact amount of light passing through the sample. The meter scale can be read directly in either transmittance or absorbance units.
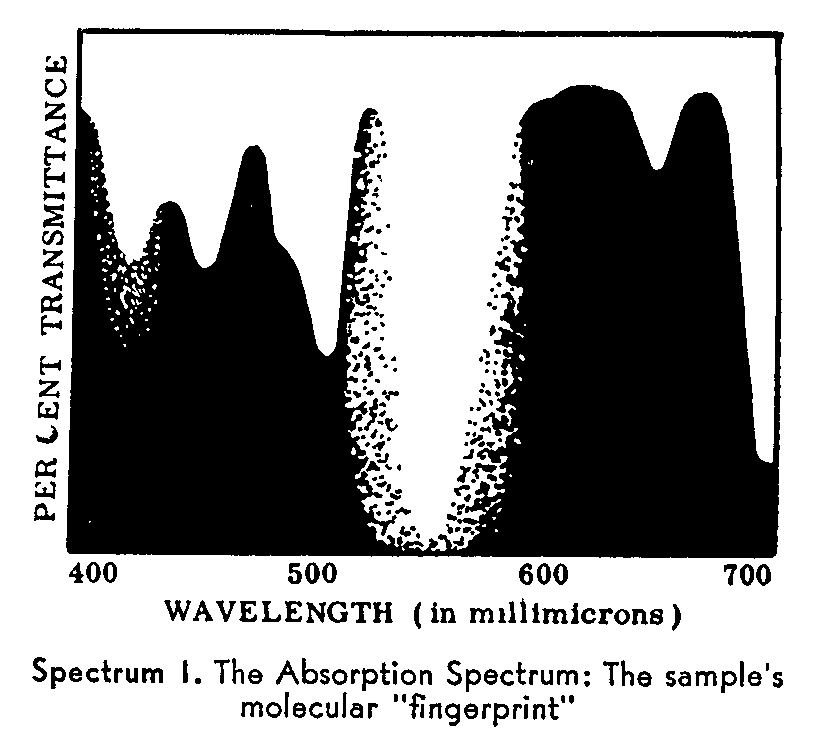
Transmission (Absorption) Measurements. The drawing above shows how the Spectrophotometer measures the amount of a particular color (wavelength) of light absorbed or transmitted by the sample substance. By making a series of such measurements and using different wavelengths of light, the analyst can draw a curve as in Spectrum I which shows the exact location and degree of the absorptions of the sample over a wide range of wavelengths. This curve is the "absorption spectrum" ... the unmistakable "fingerprint" of the sample.
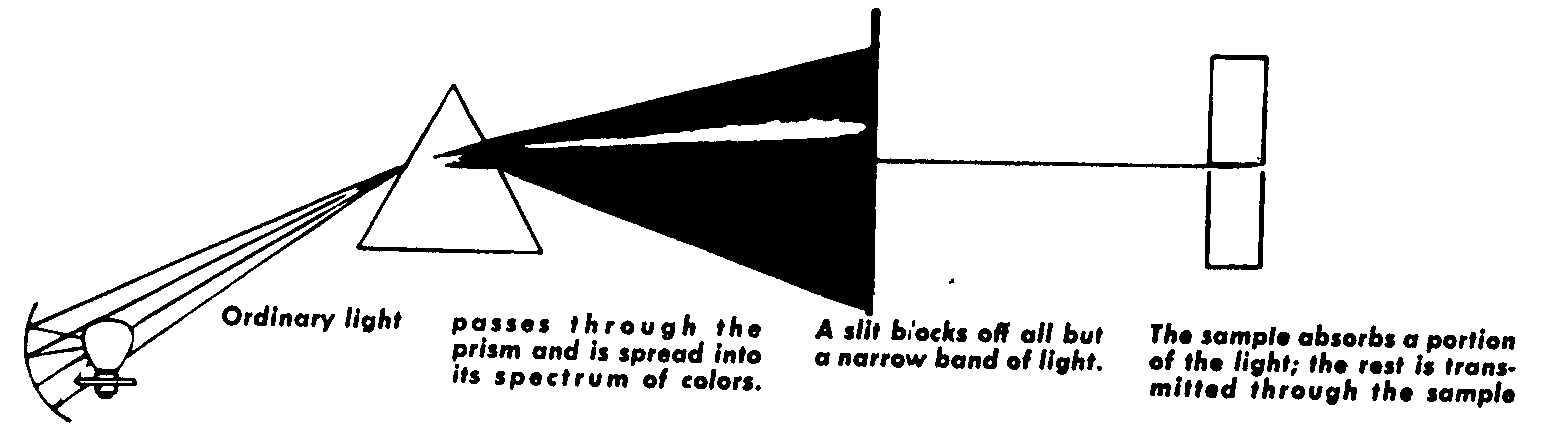
Reflection Measurements. The narrow band of color passing through the slit enters the Reflectance Attachment (below) and is directed downward onto the opaque, colored sample. The sample absorbs some of the light. The rest of the light is reflected, strikes the phototube and is registered on the meter. The "reflectance curve" (Spectrum 2 below) is the result of a series of such measurements made at different wavelengths. It is the only true picture of a color.
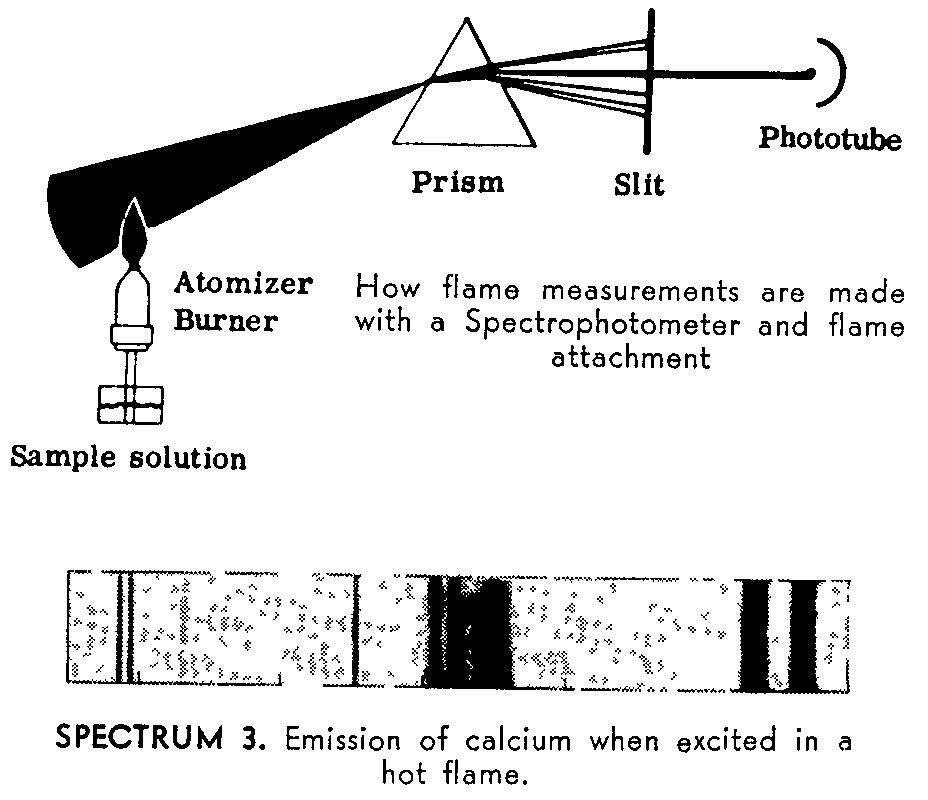
Flame Measurements. The sample, in liquid form, is atomized into a hot flame, as illustrated below, which acts as the light source for the Spectrophotometer. This flame excites atoms in the sample, causing them to emit radiations at various wavelengths (See Spectrum 3). By measuring these emissions, the Spectrophotometer enables unknown elements to be identified and amounts of known elements to be determined. Called "flame photometry", this method is extremely rapid (as fast as 10 seconds a measurement) and to date has been applied to over 50 elements and 700 substances.
Reproduced by permission of the Beckamn Instruments Inc., South Pasadema, Colifnornia, U.S.A.

Historical development of methods of recording spectra. Absorption spectra of benzene. Upper left. Benzene absorption spectra, according to W. N. Hartley, 1880, by method of relative thicknesses. Upper right. Photographic record of absorption spectra made on a Hilger spectrophotometer with tungsten steel arc, and conventional spectrograph, from Lothian. Lower left. Line graph constructed from the photograph upper right from Lothtian, 1950. Lower right. Benzene spectra produced by the Cary recording spectrophotometer by permission John Wiley & Sons and Friedel and Orchin
In certain kinds of work where it has been found sufficient to isolate only certain spectral regions, optical filters have proved to be satisfactory. These filters are of three types: ([1] ) glasses, usually two or more in combination; ([2] ) two or more dyes incorporated into gelatin or other media; and ([3] ) interference filters. Information on the filtering characteristics of these filtering media may be obtained from the Corning Glass Works ([6] ) (Glass Colour Filters (1946)), the Eastman Kodak Company ([7] ) supplying the Wratten dye-gelatin filters (Wratten Light Filters, 1948), and the Farrand Optical Company ([8] ) (Farrand Interference Filters, 1947) and Baird Associates ([9] ) (Baird Associates Interference Filters, 1946) supplying interference filters.
Spectrophotometry consists essentially of measuring the ratio of two radiant energies at a specified frequency or wavelength, and then repeating this measurement at other frequencies or wavelengths as often as desired over the spectral range of interest. This is illustrated in figure 1. This ratio of radiant energies may be determined by visual, photographic, or photoelectric means. In visual spectrophotometry the photometric part of the instrument includes a two-part photometric field, and a means of varying the luminance of one of the two parts, so that the eye is used only to detect the unmatched, and finally to judge the match of the two parts of the field. The means of varying the luminance of one part of the field is calibrated, so that the actual value of the match point is determined by an auxiliary system.
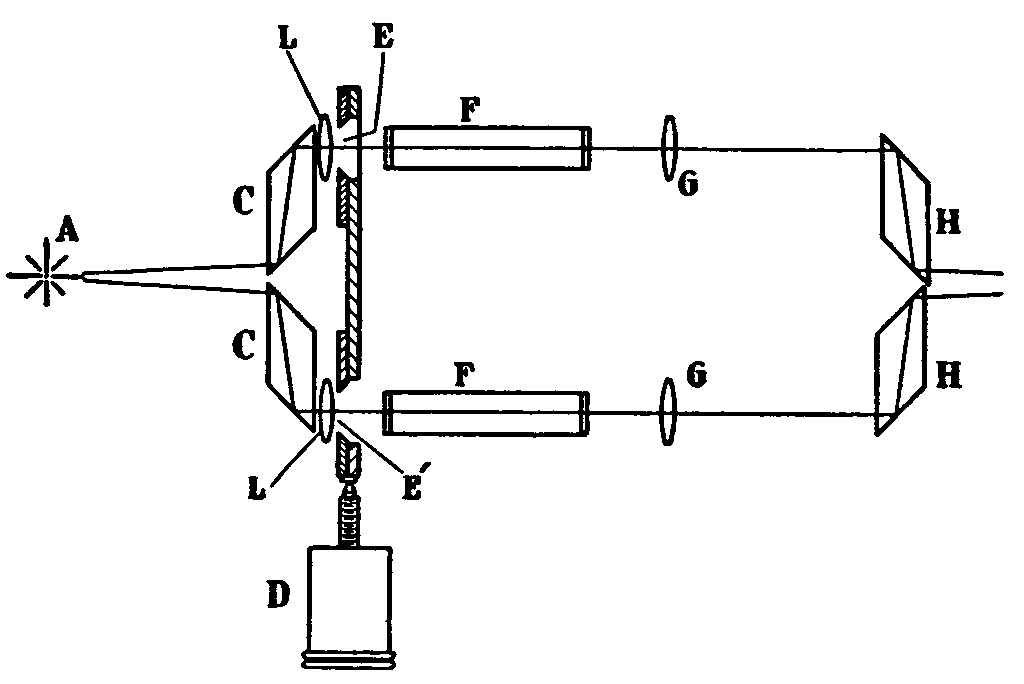
Photographic photometry is normally used when a conventional spectrograph is used as a dispersing medium. A device known as a Spekker Photometer ([10] ) (Hilger, 1937) illustrated in figure 3 is used in front of the entrance slit of the conventional spectrograph to illuminate it by two beams of light at the same origin A, the intensity of the one being varied by the absorbing medium under test, whilst the intensity of the other beam is controlled by the size of the opening of the variable aperture (E'). The angles of the prisms (H) (H') have been chosen so that the images of the source of light formed by the lenses (G) (G') are bisected at the junctions of the prisms (H) (H'), the two halves being thus brought together for comparison to be made of their intensities. In this way it is ensured that the same portion of the source of light is used for making a comparison at the dividing line of the two narrow bands of the spectrum.

The Spekker Photometer, taken from Lothian by permission of Hilger & Watts, London, England
This spekker was used to take the photograph of the benzene spectra in figure 2 (see reference [11] ).
If we consider the photograph of the emission spectra, we shall see it consists of a series of pairs of spectra in close juxtaposition, one of which is of a certain intensity, while the other, namely, that which has passed through the material under test, is more dense than that which has passed through the solvent, in certain parts, and less so in others, there being definite portions of the spectrum where the intensities of the two are equal.
Considering the points of equality, the equal blackening of the photographic plate indicates that the light passing through the test material is equal to that which has passed through the variable aperture (E') (figure 2). When no absorbing material is present the spectra are of equal intensity only when the areas of the fixed and variable apertures are the same, the ratios of the respective areas being a measure of the ratio of the intensities of the two beams of light.
Returning to the points of equality of blackening of the photographic plate in figure 1, one can say that the intensity of the light after passing through the medium under test is equal to that passing through the variable aperture.
The apertures' areas are proportional to the light transmitted, and at points of equality of blackening, the area of the variable aperture has been adjusted to make the intensity of the control beam equal to that of the light passing through the medium; the ratio of intensities may be represented by
|
Io |
Area of fixed aperture | |
|
-- |
= |
------------------------- |
|
I |
Area of variable aperture. |
The drum (D) (figure 3) which controls the variable aperture is engraved to read Log-Io/I , which logarithm is usually referred to as the "density".
By taking a series of photographs with the drum set to different values of density and searching out points of equality of the two spectra (points marked with a dot in photograph of spectra in figure 2), one is able to obtain a number of values of Io/I for a wide range of wave-lengths, and by connecting up the dots so obtained one can draw a graph connecting density (i.e., log Io/I) with the wave-length of light. These values are plotted as extinction coefficient (optical density) on the graph lower right hand corner figure 2. Because of the higher dispersion normally associated with spectrographs more fine structure may be investigated by this technique than can be observed visually or with photoelectric photometers.
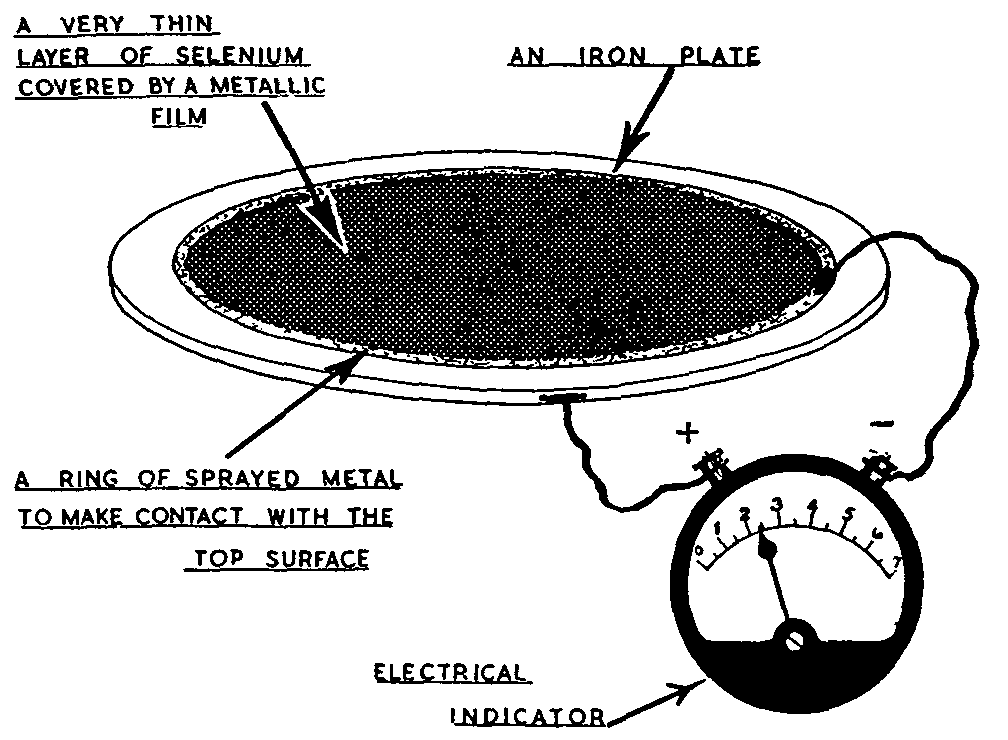
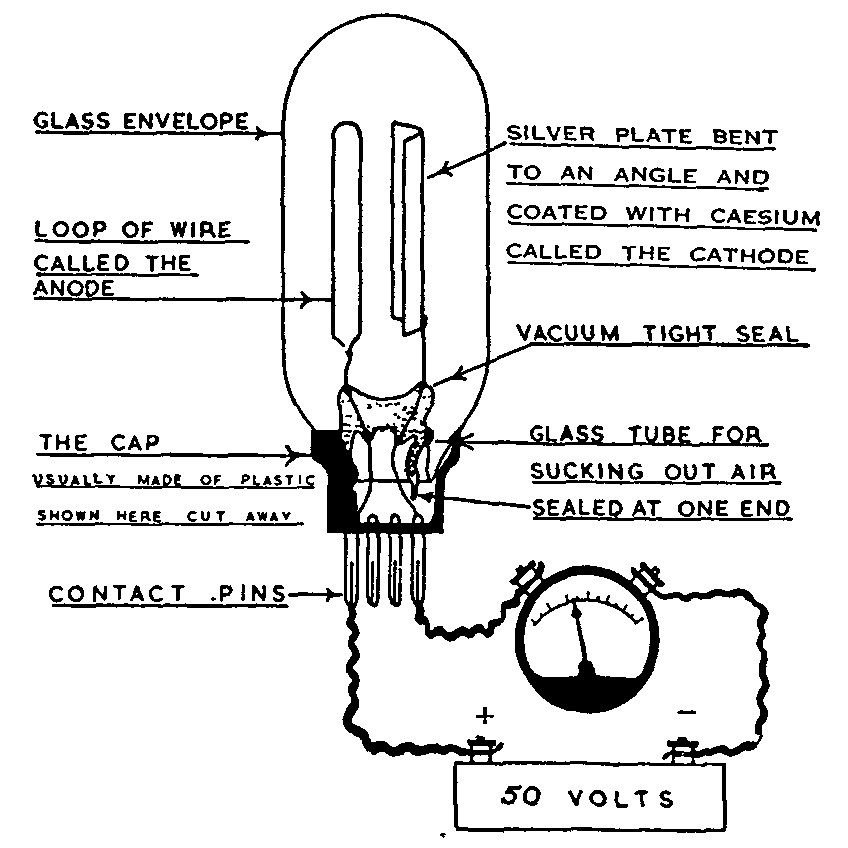
Photoelectric detectors are of three types: ([1] ) barrier layer cells (figure 4); ([2] ) phototubes; and ([3] ) photomultiplier tubes (figures 5 and 5a). A barrier layer cell ([12] ) consists essentially of a plate of either copper or iron upon which a semi-conducting layer of cuprous oxide or selenium has been grown. The semi-conducting layer has been covered with a light transparent layer of gold, platinum, copper, or lead which acts as an electron collector. The average cell has an output of about 120 microamperes per lumen and can be used in conjunction with electrical indicators such as micro or milliammeters to detect and measure high levels of illumination. No external potential source is required with a barrier layer cell. Vacuum phototubes ([12] ) require a source of potential illustrated in figures 5 and 5a and are normally used with a vacuum tube amplifier as shown in figure 7.
Over the range of intensities of radiation encountered in spectrophotometry the photocell is designed to give a linear response. Radiant energy comprised of photons dislodges electrons from atoms in the cell surface, which are drawn away from the surface by an electric potential producing a continuous electric current. The number of electrons which leave the surface is dependent on the amount of radiation falling upon it; the greater the number of electrons which come out of the surface the greater is the current produced. All photometers are designed on the basis of the linearity of this photoelectric response. For a linear photocell, R=Kα I, where R=instrument reading, I=intensity, and Kα=constant. Then Ro = Kα
|
|
Ro |
|
Kα Io |
|
|
Io and log 10 |
R |
=log 10 |
Kα I |
true absorbence. Not all |
photocells are linear in response to radiation according to Cannon and Butterworth (1953). A non linear spectrophotometer, although giving a linear Beer's law plot, will give erroneous values for absorptivity.
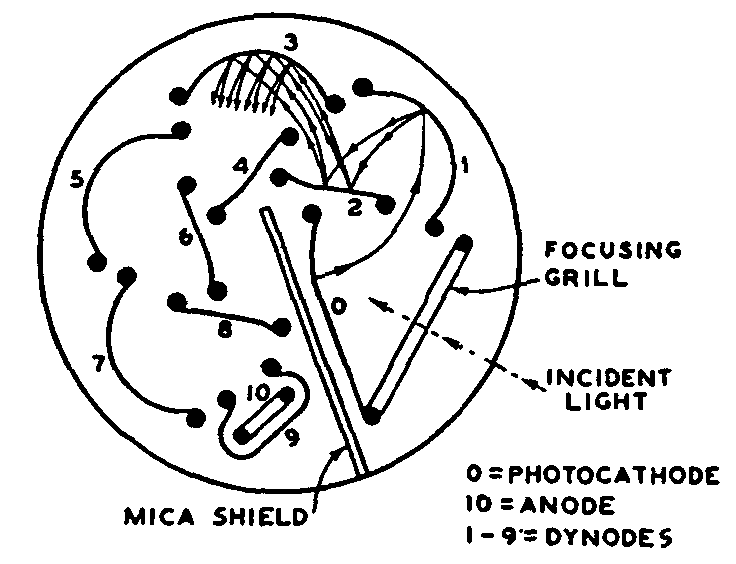
A vacuum tube of a special kind employing several electrodes or dynodes which achieves the equivalence of amplification within the tube itself is called a photomultiplier tube. These tubes have a higher signal-to-noise ratio than the phototube amplifier combination. The IP28 multiplier phototube (figure 5a) is capable of multiplying feeble photoelectric current produced at the cathode by an average value of 1,000,000 times when operated at 100 volts per stage. The output current of the IP28 is a linear function of the exciting energy under normal operating conditions.
The Beckman spectrophotometer is the instrument currently most often used in North America for measurement of absorption spectra. It is designed to operate in the ultraviolet, visible, and with some mechanical adjustments in the near infrared regions. A description of its three main parts, monochromator, photometer, and amplifier, will indicate how the spectrophotometer works.
The optics of the Beckman spectrophotometer are shown in figure 6. One of the two interchangeable radiation sources is used at A, depending on the spectral range desired. A standard 32-cp. prefocused tungsten lamp, (Mazda 2331) is used for the spectral region from 320 to 2,000 mμ. A special hydrogen discharge tube is used for the spectral region from 200 to 320 mμ. A 6-V. heavy-duty storage battery serves as the power source for the former. A new development which is useful when contemplating continuous use of the instrument over a long period is the Beckman D.U. power charger. This accessory has a space for the 6-V. battery and also locates the C batteries outside of the monochromator case. A special power unit or "powerpack" (as it is sometimes called) operates the hydrogen discharge tube. The two sources are mounted in a single housing on separate backs for ready changeover. A mercury discharge tube is also available to check the wave-length setting of a number of known wave-lengths.
The Barrier Layer Photocell, from the publication Electric Eyes, by permission of the Tintometer Ltd., Salisbury, England
The Phototube, from the publication Electric Eyes, by permission of the Tintometer Ltd., Salisbury, England
The Multiplier Phototube
The optics of the Beckman spectrophotometer from the publication Electric Eyes, by permission of the Tintometer Ltd, Salisbury, England
The wiring diagram of the Beckman phototube amplifier, from Kirk
The control panel of the Beckman spectrophotometer, from Kirk
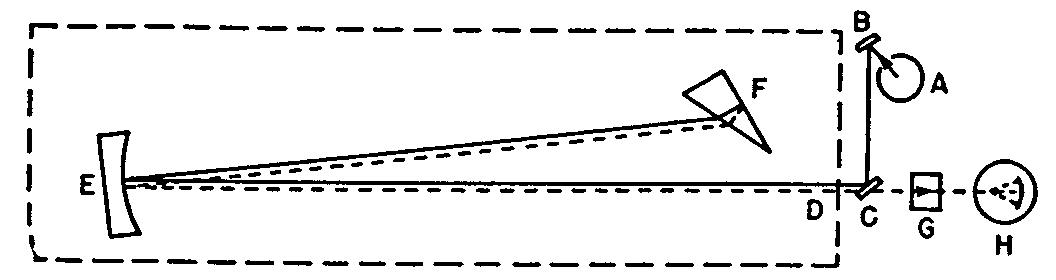
Radiation from source A is magnified by the collimating mirror B, and the diagonal mirror C, and is focused on the plane of the slit at D. After passing through the slit opening, the radiation is reflected by the collimating mirror, E, onto the prism, F. The back surface of the prism is an aluminized mirror, and the radiation is reflected by this mirror, back through the prism and the mirror E to the slit D.
The internal optics, mirror E, and prism F, are so arranged that the emergent beam is approximately half an inch above the entering beam at the slit, D. The emergent beam then passes through the absorption cell in the cell holder, G and into the photocell, H.
The quartz prism, F, was selected as the light dispersion (splitting) device because it gives a spectrum relatively free of scattered light. The half prism, F, and reflecting mirror, E, not only require less quartz than a full prism but also obviate several complications which would arise if a full prism were used. Likewise, the use of collimating mirrors avoids the necessity of quartz lenses for focusing the radiation.
Variation in wave-length of the radiation is obtained by rotating the prism with a simple mechanical device. The wave-length setting can be varied slightly by moving the position of the collimating mirror, E; in actual practice this is only used in making initial adjustments.
The output (voltage) of the photocells is amplified by an electronic amplifier whose circuit is shown in figure 7. The essential feature of the circuit is that the phototube current is measured by balancing the voltage drop in the 2,000-megohm resistor (R10) with the slide wire potentiometer (R1). The indicating device is a sensitive milliammeter. The remaining controls are included to correct for stray currents in the amplifying circuit, and to adjust the circuit so that the percentage transmission and/or absorbence is read. The absorbence A (sometimes called optical density) is also read directly on the same scale as the transmission.
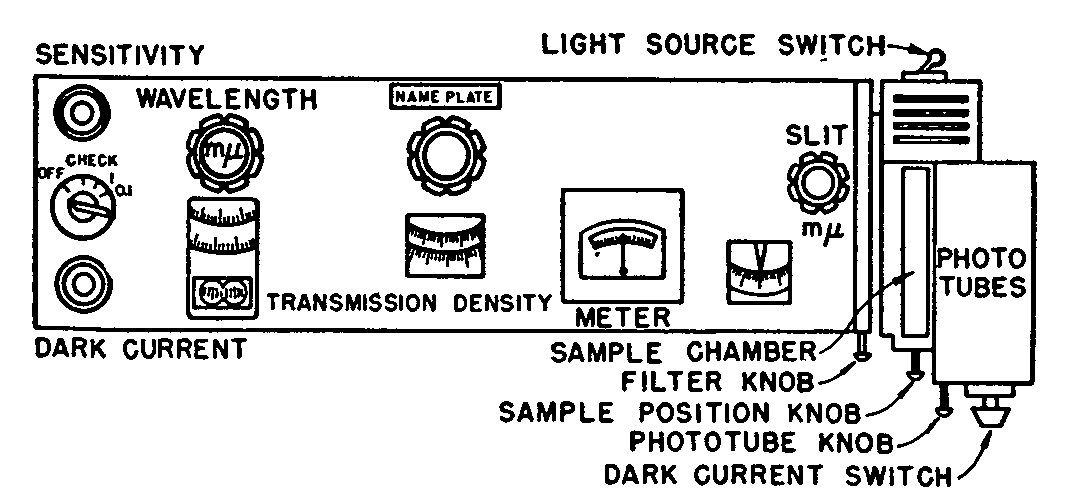
The various knobs and dials on the instrument are shown diagrammatically in figure 8. The wave-length knob controls the wave-length of the radiation passing through the cell holder (solution) and to the photocell. The wave-length scale indicates the radiation wave-length being isolated. The scale is not evenly divided and at one end (250-300 m μ) the scale divisions appear closely crowded together. The per cent transmittance (or absorbence) knob and scale are used respectively to balance the meter when a measurement is being made, and to indicate the percentage transmission and absorbence reading. Two scales are available, the upper being percentage transmission; the lower scale being absorbence (previously called optical density). For narcotic identification work the absorbence scale is used; for routine quantitative work the transmittance scale is often preferred. There appears to be a greater tendency to use absorbence values in all recent work. The third of the large control knobs adjusts the width of the slits.
The amplifier system of the spectrophotometer and the measurement of the absorption or transmission of an object requires that the output of the photocell when amplified gives a constant current. Since the outputs both of the source and the phototube vary with the wavelength of the radiant energy, some device must be inserted into the circuit to control either the output of the source, or the photocell, or both. In the Beckman instrument, both outputs are controlled. The slits at D in figure 6, operated by the third big knob on the panel, control mechanically the output of source A. A rheostat marked sensitivity varies the output of the photo-tube electrically. In practice, the two adjustments are used together, the slit width (large knob 3) being a rough adjustment, and the sensitivity rheostat a fine adjustment.
In addition to increasing the amount of radiation reaching the phototube, increasing the slit width widens the dispersion range of the radiation. This dispersion range should be kept as small as possible for accurate work; the slit width should be kept as low as possible. The dial setting is usually between 0.01 and 0.06 mm. for quantitative analytical work. (In practice, when obtaining a point by point description of the spectral curve the slit width setting varies with each wave-length, being wider for wave-lengths where radiation intensity is low; the slit widths therefore may vary from 0.06 mm. to 2 mm.)
The ammeter between the transmission and slit width knobs serves only to indicate a zero or "null" point, and the several divisions on either side are for convenience only. Throughout the various adjustments of the instrument, the object is to keep the needle at the zeropoint (i.e., constant current).
On the extreme left end of the instrument are two resistances and the main switch. The dark current resistance balances out any stray current in the amplifier. The sensitivity control is a potentiometer with a 9-turn helical winding and is essentially a fine adjustment whose purpose is the same as the slit adjustment, i.e., to control the output of the photocell by reducing the current electrically. The instrument should be operated with this control approximately from one to four turns from its most counter-clockwise position, which is its most sensitive range. Operation at the other end of the sensitivity scale, i.e., the clockwise end, requires a wider slit opening, and makes the response to the ammeter extremely sensitive to changes in the position of the transmission-absorbence knob.
The main switch has three "on" positions marked "Check", "1", and "0.1". The "1" position is the true "on" position. The "check" position is used when it is desired to adjust the meter against the blank. It avoids the necessity for returning the absorbence to zero, or transmission scale to 100, before each measurement. For readings greater than an absorbence of 1 (or less than 10 per cent transmission), the switch is turned to the "0.1" position, and the meter is balanced in the usual manner. The observed transmission scale reading is then divided by ten to give the true transmission. The absorbence reading is also changed. The value 1 is added to the observed absorbence reading. This permits a more accurate measurement on the scale in the region below an absorbence of 1.
On the right hand end of the instrument are three push-pull knobs and a switch. The first of the three knobs controls a filter which is necessary when measurements are made below 400 mμ with the tungsten light source. When the knob is in as far as possible, there is no filter in the light path. With the knob pulled out into the next position, a purple filter is introduced into the light path. This cuts off stray radiation which may enter the monochromator when the above combination of source and wave-length is used. The third and last position is blank.
The second knob (at the right hand end) controls the position of the absorption cells in the cell holder. This obviates the necessity of opening the cell container while a series of measurements is being made.
The third knob is a switch which selects the photo-tube and automatically makes the necessary circuit changes. No one phototube can cover the entire spectral range of the Beckman instrument. Two tubes are employed which are interchanged merely by pushing the phototube knob. When the knob is furthest out, the ultraviolet sensitive tube is in position, and all measurements below 620 mμ should be made with this phototube. For measurements between 620 and 2,000 mμ, the red sensitive tube is put in position by pushing the phototube knob in as far as possible.
The off-position of the "off-on" switch shuts off the photocell both optically and electrically from the remainder of the instrument. It is put in the off-position only when the dark current is being checked.
|
Spectral regions, mμ |
Filter control |
Phototube control |
Source |
|---|---|---|---|
|
220-320 |
In |
Out |
Hydrogen |
|
320-400 |
Out |
Out |
Tungsten |
|
400-620 |
In |
Out |
Tungsten |
|
620-2,000 |
In |
In |
Tungsten |
Qualitative absorption spectrophotometry may be achieved by comparing the absorption curve of the unknown drug with curves of known compounds. Accurate data on the absorption of a sample as a function of wavelength must be obtained over a considerable portion of the spectrum to allow this method to be used. Similar structures within different molecules will show similar absorptions and likewise, very similar complex organic molecules will show very similar absorption spectra. In fact this generalization is so well established that tables of the wave-lengths of the absorptions of different groups in the ultraviolet and visible regions have been made ([14] ) (see Harrison, Lord and Loofbourow, 1948).
Unfortunately no single comprehensive compilation of absorption spectra exists; some data have been compiled by the American Petroleum Institute Research Project 44 at the National Bureau of Standards. International Critical Tables ([15] ) contains both absorption data and references to literature containing spectra of both organic and inorganic compounds. Tables annuelles de constantes et données numériques, données numériques de spectroscopie, vols. I to XII by V. Henri and L. Bruninghaus ([16] ) contain tabulations of absorption data. Literature references to absorption are contained in Physikalische Chemische Tabellen, edited by Roth and Scheel ([17] ); Handbuch der Organischen Chemie, fourth edition by Beilstein ([18] ); and Handbuch der Inorganischen Chemie, eighth edition by Gmelin ([19] ).*
Sources of information including books, review articles, miscellaneous articles, instrumentation and technique, and manufacturers, were collected by Hirt (1952) ([20] ). Hirt is also in charge of a project for the publication of UV spectra. A recent atlas of ultraviolet spectra of aromatic compounds was written by Friedel and Orchin (1951) ([21] ). Since no critical survey of the literature of absorption spectrometry has been made with respect to narcotics, a more or less complete historical review of ultraviolet absorption spectrophotometry related to narcotic analysis is included in the next section.
The earliest reference ([022] ) to the study of ultraviolet spectra is by Brewster (1833) although Miller ([023] ) and Stokes (1852) appear to have done the first serious work ([024] ). Sir William Noel Hartley (1881), Chemistry Professor, Royal College of Science, Dublin, using Miller's original quartz spectrograph ([023] ) reported the first ultraviolet spectra of narcotics, and he appears also to have been the first to suggest the use of these spectra as evidence in alkaloid identification in court cases. In a report ([025] ) to the Royal Society of London, Dec. 1884, Hartley described the results of measurements and illustrated by means of graphs the spectra of thirty-two alkaloids, including the following opium bases and their derivatives: morphine, narcotine, codeine, the-baine, papaverine, oxynarcotine, apomorphine hydrochloride, cotarnine hydrobromide, diacetylmorphine and acetylcodeine.1Keeping in mind the relative knowledge of the time, the comments of Hartley are interesting. The difficulty of identifying these alkaloids by distinctive chemical reactions and their physiological action was pointed out by Hartley. "The facts", he stated in 1884, "are well known, and are generally made use of by counsel for the defence in medico-legal cases ...." and further as an example, Hartley states that "The evidence given at the trial of George Henry Lamson, a surgeon, at the Central Court in 1882, for poisoning with aconitine, conferred greatly increased importance upon any method of absolute physical measurement which might be substituted for the ordinary tests in the identification of the dangerous alkaloids. Almost all alkaloids have a complex chemical constitution, and every complex molecule in which carbon is in a certain state of condensation has a definite absorption spectrum in the ultraviolet if not in the visible region. In most cases the absorption curve is peculiar and often strikingly characteristic, and only minute quantities of material are actually required for obtaining measurements of spectra from which such curves may be plotted. The interest that attaches to an examination of the absorption spectra of the alkaloids is not alone the fact that a means of recognizing, detecting, and estimating such substances may be devised, but still more, that we may learn something of their chemical constitution."
Project No. El3 of the American Society for Testing Materials for the compilation of spectral data includes IBM coding. Inquiries should be directed to Dr. R. C. Hirt (see reference [20] ) or Dr. George Buc, editor of Applied Spectroscopy.
Hartley's conclusions (1884) also showed his main interest to be in structure determinations, for he indicated that morphine, codeine, and their derivatives apomorphine hydrochloride, mono-acetylmorphine, and diacetylmorphine,2all have a similar condensed nucleus. This nucleus Hartley believed was a benzene or pyridine derivative, since as he showed, the main absorption band extended from 2,600 ? to 3,000 ?. He showed, also, the effect of alkyl and acetyl substitutions, upon the curve of absorption as exemplified by codeine and the acetylated derivatives of codeine and morphine. Later in 1887 Hartley from these and other studies advanced the belief that the absorption spectra of derivatives are characteristic of the parent molecule of an organic substance and are merely altered by additional chromophores, auxochromes, or substituents ([26] ).
Dobbie became a student of Hartley's in 1898 and did further work in measuring spectra, some of which were of narcotics. In 1903 Dobbie measured the spectra of cotarnine and a number of alkaloids ([27] ) and discussed their molecular constitution in terms of spectra. Dobbie also measured the spectrum of papaverine. He used the tautomeric equilibrium reaction of cotarnine in basic solutions as an indicator of the strength of alkaline hydroxides, and followed the course of reaction by means of ultraviolet spectrophotometry. In 1911 Dobbie showed the identity of the spectra of codeine and neopine ([27] ); and in 1914 related the structure of a number of spectra of isoquinolines with that of papaverine.
1The last two were called tetraacetylmorphine and diacetylcodeine, at the time.
2Which he called diacetylmorphine and tetraacetylmorphine.
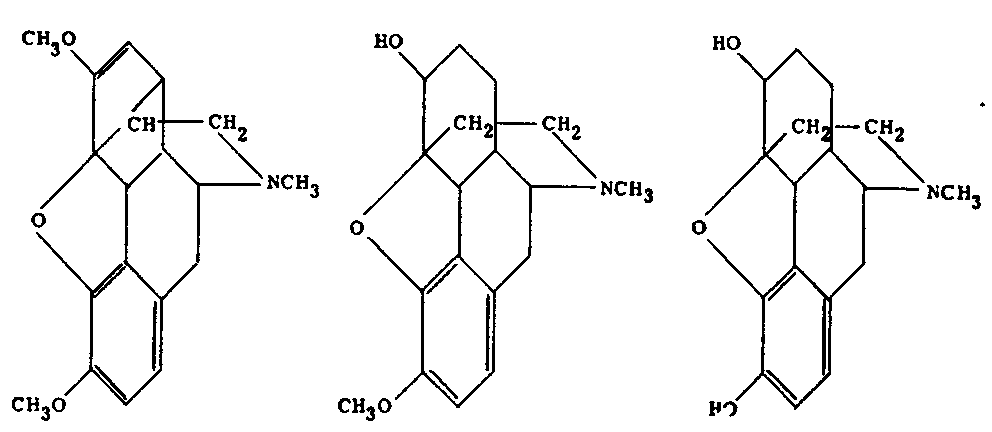
Left: Morphine Scale of wave lengths
Middle: Codeine ([2] ) Scale of wave lengths
Right: Thebaine Scale of wave lengths
The line transmission of spectra of morphine, codeine, and thebaine, according to Hartley (1885)
Lothian (1937) critically evaluated this early work and made the following comments about it ([11] ).
"Unfortunately, much of the work done prior to about 1910 was valueless, since the methods employed were either entirely qualitative, or at best semi-quantitative, compared with those available today. Some uniformity was introduced however, by Hartley and other workers after him who developed a method in which the wave-lengths at which various thicknesses of a given solution ceased to transmit were recorded photographically" (see ordinate scales, figures 2, 9 and 13). "By diluting the solution progressively, the course of the absorption could be followed up to its maximum of intensity, and a curve of equivalent thicknesses of standard solution (or logarithms of these thicknesses) as ordinates against the observed limiting wave-lengths transmitted as abscissae then reproduced the main characteristics of the absorption band." ([44] )
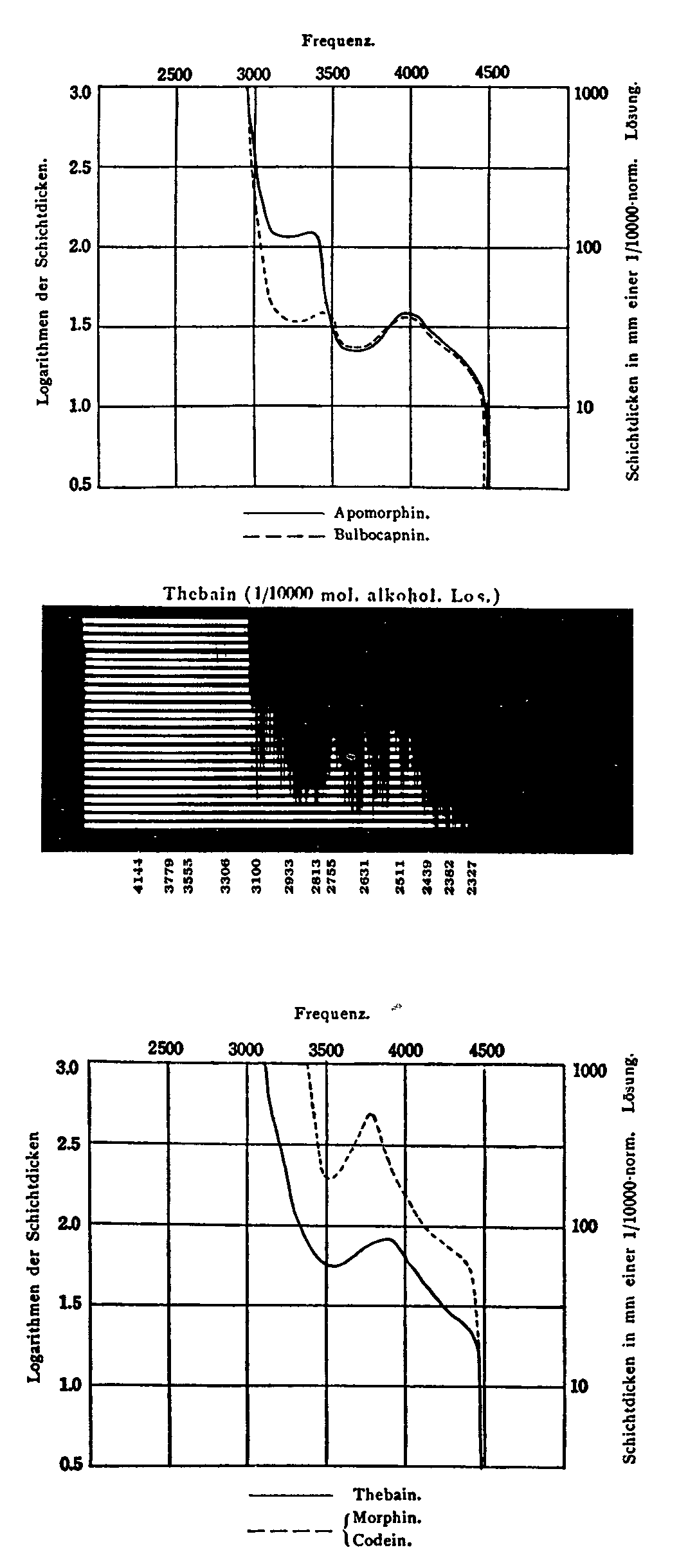
The absorption spectra of morphine, codeine, and the-baine determined by this "equivalent thickness method" as recorded by Hartley ([25] ) (figure 9) are compared with the same spectra shown in figure 10 which were obtained by a modern photometric method and plotted in terms of logarithm of the absorbence index. The location of maximum of absorption spectra by Hartley seem to be within 5 m μof our values, but the intensity parameter determined by the equivalent thickness method does not show the relation to quantity used in Hartley's experiments and unless the conditions of measurement were identical, the curves for different substances, morphine, codeine, and thebaine, would not be strictly comparable. The use of the absorbence index ( ε) makes such comparisons possible. Another point is that the detailed course of absorption (fine structure) is not recorded by the older method as can be seen by comparing the sequence of benzene absorption spectra (figure 2) recorded by Hartley (1883) and by Friedel and Orchin (1950) using a Cary recording spectrophotometer ([21] ). This sequence indicates the historical development of ultraviolet spectra starting with the plotted record of perhaps the first practical quartz spectrograph (1885), through the photographic era with the Hilger Spekker ultraviolet photometer and medium quartz spectrograph (1910-1940) to the continuous 10 minute record of the Cary spectrophotometer (1950). The difference in fine structure and range may be seen in this comparison.
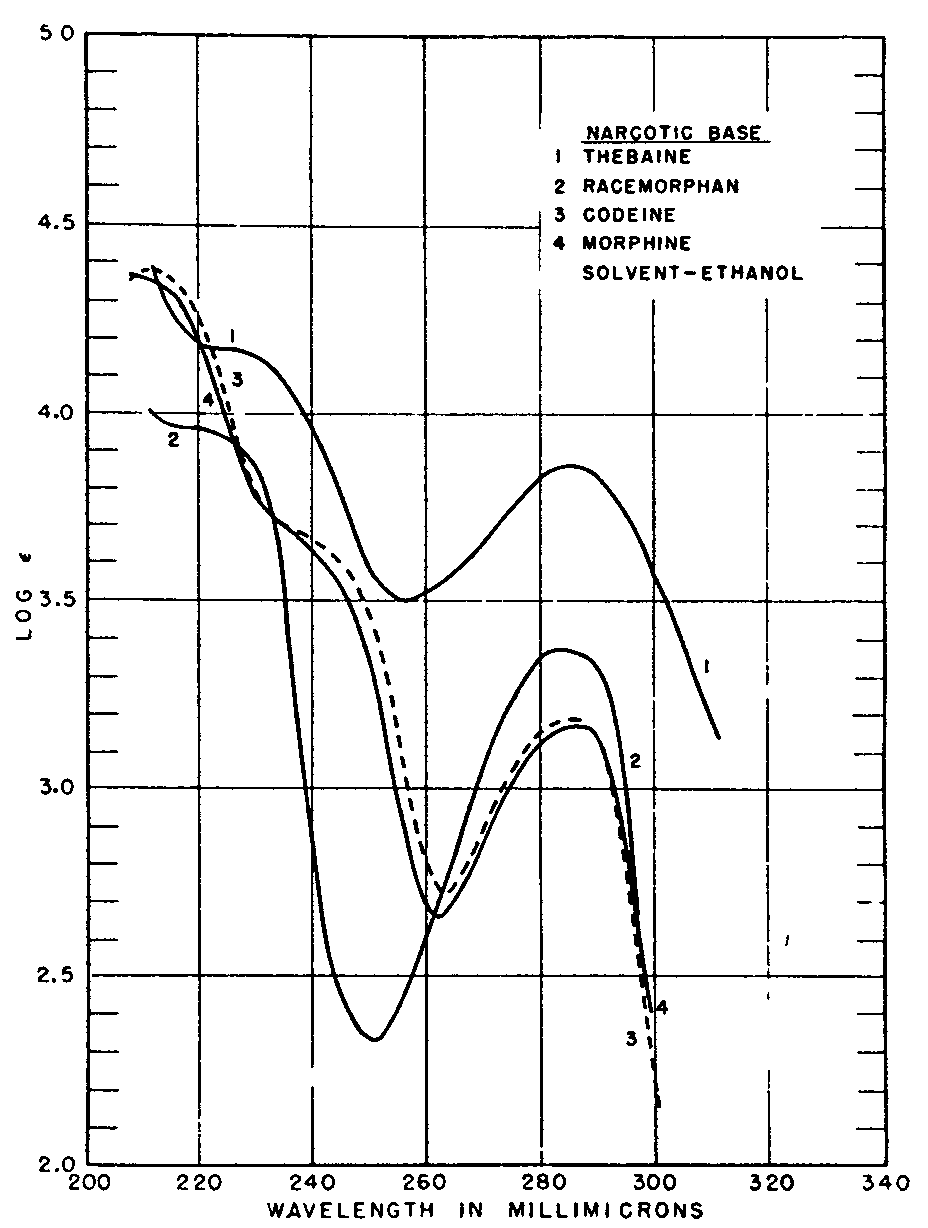
Comparison of line, absorption, spectra of morphine, codeine, thebaine, and racemorphan by a modern spectrophotometer
Henri (1913) ([28] ) introduced a new concept into absorption spectrophotometry which enabled him to plot the intensity in terms of wave-length in a new way. Schwarzschild had discovered that the blackening of a photographic plate is related to the quantity of light falling on it by the expression
density of blackening +/- I tn
where n is a constant of the photographic plate, and method of development. I is the intensity or quantity of light, and t is the time of exposure. Henri applied this relationship to the determination of the ratio of intensities of light passing through a solution containing a light absorbing substance, and through the solvent itself.
If spectra are photographed through equal lengths of solvent, l for a time t o; and through the solution for time t, there will be wave-lengths at which the density of blackening will be equal in each spectrum. Then at these wave-lengths if I o and I are the respective transmitted intensities
I ot o n= I t n
I is that part of the incident intensity which would be transmitted by the solute alone, so that
density=log 10 Io/I=n log 10(t/t o)
The practical application of Henri's density wave-length relationship required the independent determination of n. Henri was the first to use the logarithm molecular extinction coefficient plotted against wave-length. Gompel and Henri (1913) ([28] ) studied the ultraviolet absorption of morphine, codeine, and apomorphine and compared them with phenanthrene. They utilized the expression
|
|
IoI |
|
|
ε== log |
I |
(cm. mol.) |
and plotted the log. ε against wave-length λ in angstrom units. This symbol ε, called extinction, probably derives from the fact that the measurements are made at the points of equal extinction on the photographic plate. The spectra of absorption for phenanthrene, apomorphine, morphine and codeine by Henri are shown in figure 11). Henri's morphine (codeine) spectrum shows a maximum at 2,830 ?.
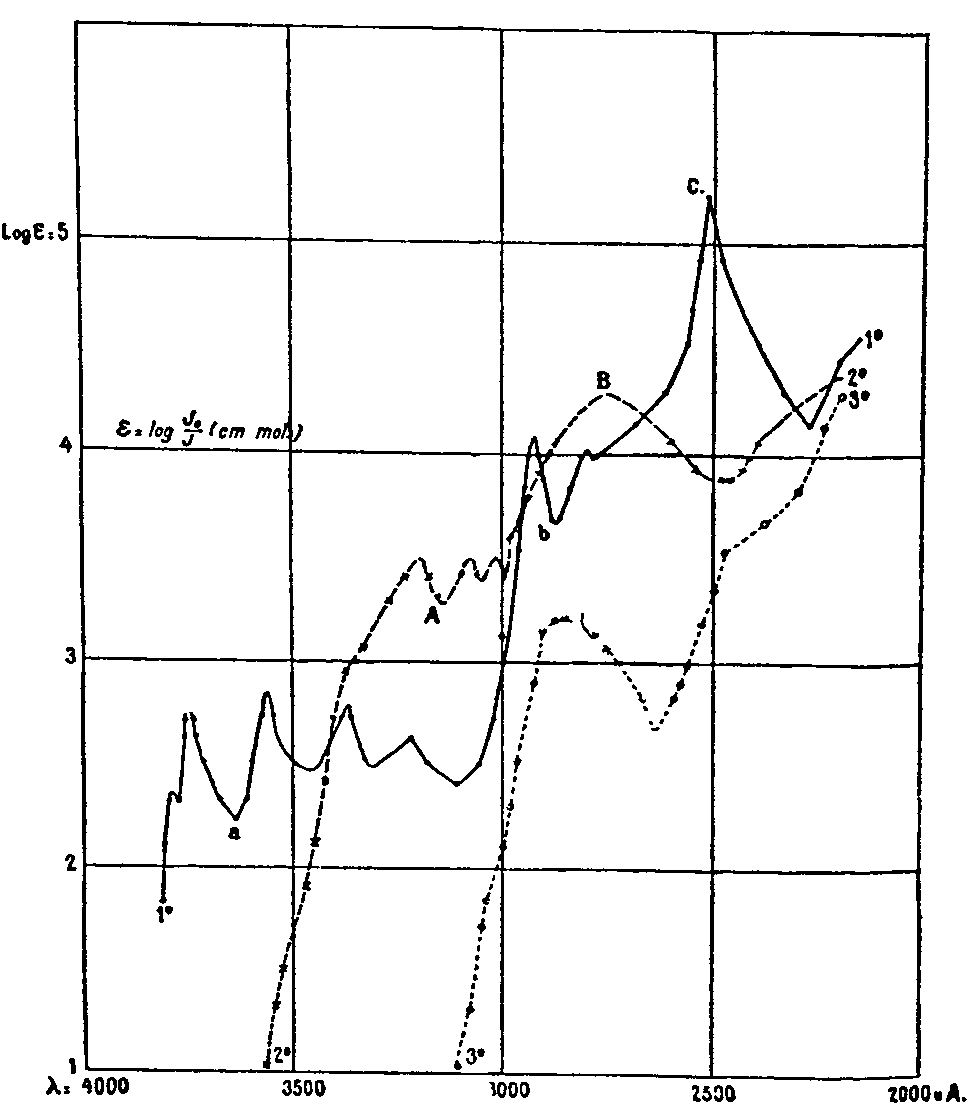
Spectrophotometric absorption of phenanthrene, apomorphine, morphine, and codeine, according to V. Henri (1913)
Henri ([29] ) ([51] ) ([52] ) carried further the chromophore concept earlier stated by Hartley, i.e., that the absorption spectrum does not result from the action of the whole molecule on the incident radiation, but as a result of only particular parts of a molecule. For example, Henri showed that simple ketones exhibit an absorption band with an absorbence index of about 15 between 2,700-3,000 ?, acetone having a maximum at 2,800 ?. Using some examples from our own experience with narcotics it is observed that complex ketones like dihydrocodeinone, dihydromorphinone, dihydrocodeinone enol acetate, ketobemidone, methyldihydromorphinone all have an absorption maximum at about the same location as the acetone maximum, namely at 280-282 m μ. With the exception of ketobemidone they all have extinction coefficients (E=K C d) of about 1,200 to 1,250. Ketones belonging to the diarylalkoneamine group like amidone, isoamidone, pipidone and phenadoxone, all absorb between 292-296 m μ, at much lower intensities (about ε =500). Henri's observations on such similar homologous series of ketones were: ([1] ) that the absorption curves were similar in shape, that the main maximum moves toward longer wave-lengths and the intensity of the main band increases as the molecular weight increases. Henri found that other groups beside the carbonyl (C=O) behave in the same manner, in relation to their effect on the absorption spectra of the compound containing them. Thus Henri extended the chromophore concept to include groups other than those associated with visible colour which could give rise to a characteristic absorption in the ultraviolet region of the spectrum.
Henri and Zangger in 1912 discussed the application of ultraviolet spectra in the solution of problems of identification and detection in the field of forensic medicine ([30] ). Zangger referred to the early application of the quartz spectrograph (Hilger) in the qualitative determination of alkaloids in this field ([31] ).
Steiner (1922) studied the absorption spectra of isoquinoline alkaloids, especially papaverine and related spectra which he compared to veratrol ([32] ). The values for papaverine base and hydrochloride Steiner obtained are compared with those obtained by us. Steiner's values are listed on the left and ours on the right. Values for papaverine hydrochloride were also given by Steiner. Narceine was found by Steiner to have a maximum at 2,707 ? and ε= 9,800; we have found the same compound to have the following values, λ max=2,700 ? and ε= 9,620. Narcotine, hydrastine, opianic acid and hydro-cotarnine were also studied by Steiner. The spectrum of narcotine was the result of contributions from opianic acid and hydrocotarnine mainly determined by the benzene chromophore present in opianic acid.
|
λmax, ? |
εmax |
λmax, ? |
εmax |
|---|---|---|---|
| 3,261 | 5,400 | 3,270 | 4,720 |
| 3,117 | 4,800 | 3,140 | 3,990 |
| 2,802 | 6,400 | 2,795 | 7,170 |
| 2,346 | 54,000 | 2,390 | 68,200 |
Hans Fischer (1925) a student of V. Henri and H. Zangger at the Institute of Forensic Medicine in the University of Zurich prepared a thesis which reviewed the history and application of spectrographic methods in general to identify alkaloids and compounds of toxicological interest ([33] ). Fischer used a Hilger spectrograph, of the same kind employed by V. Henri, having a high dispersion power and yielding a spectrum measuring 27 cm. between the wave-lengths 2,200 to 3,200 ? with a precision of 0,0001 ? in this region. Fischer's opinion of the usefulness of spectrophotometry in forensic medicine was very enthusiastic. He believed that impurities play a minor role in spectrophotometric analysis. Comparing colour, crystal, and spectrophotometric tests, Fischer stated that the first two methods achieve results by a process of destruction, which in courts of law is not desirable; while spectrophotometry left the original molecule unchanged and at the same time identified the molecule. The same arguments are in vogue today by protagonists of the two schools; some recommend chemical tests, some physical tests. The answer probably is that both tests are extremely useful. A very practical demonstration of the interrelationship of physical and chemical methods is to be seen in the laboratories of the New York State Racing Commission ([034] ), where upwards of 300 samples a day can be tested employing all three types of tests in each drug analysis.
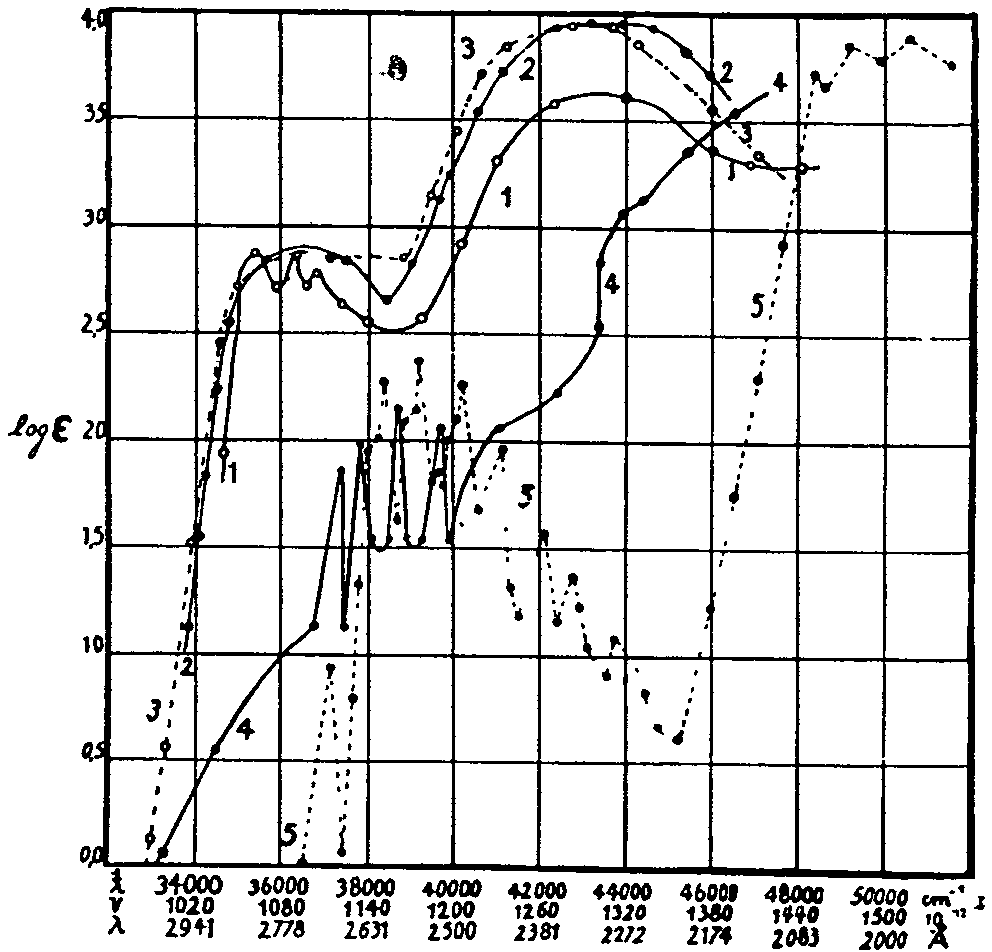
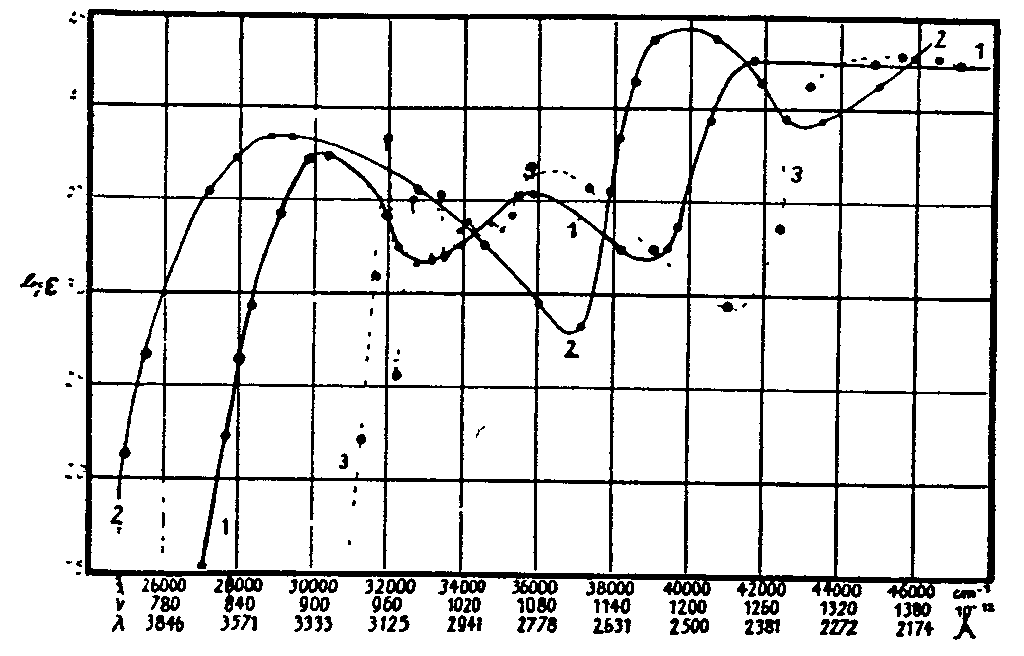
The spectra of narcotics and derivatives studied by Fischer were: benzoylecgonine, opianic acid, meconin, ecgonine, cocaine, papaverine, narcotine, narceine, morphine, apomorphine, codeine, thebaine, and heroin. In figure 12 a set of spectral curves obtained by Fischer for benzoic acid, benzoylecgonine, and cocaine are given, along with those for tropic acid and benzene. The spectra are plotted as log. E against wave-length, A, wave numbers, cm -1, and frequency 10 -12. The similarity of the absorption curves for the first three compounds is striking.
1) Benzoic acid in hexane, (2) Benzoyl ecgonine in water, (3) Cocaine hydrochloride in water, (4) Tropic acid in alcohol, (5) Benzene in hexane. Absorption curves, according to Fischer (1925)
Spectra of opium alkaloids, according to Kitasato (1927)
Bontempi (1925) reported that morphine and morphine hydrochloride had the same spectra and concluded that hydrochloric acid had no effect on the spectrum of morphine ([35] ). Eisenbrand (1926) reported that morphine could be determined in amounts as low as 0.2 mg by means of absorption spectra ([36] ).
Brustier (1926) measured the line spectra of narcotine, morphine and ethylmorphine, and expressed these graphically (37).
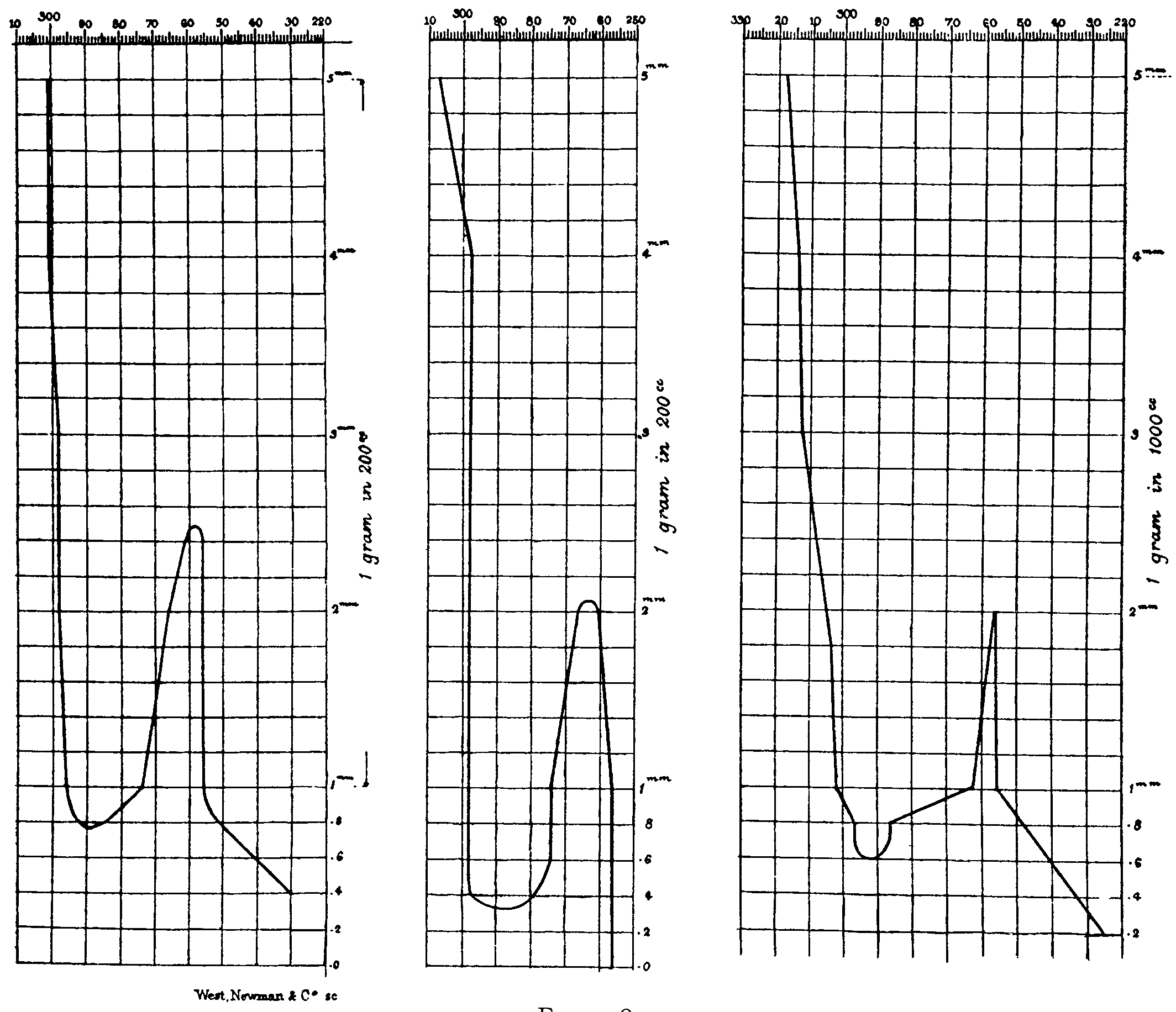
Kitasato (1927), using a Hilger quartz spectrograph, photographed the absorption spectra of 42 alkaloids ([38] ). The curves and photograph in figure 13 illustrate Kitasato's method and results. Data from the photograph of spectra of 0.001 M solution of thebaine in alcohol was plotted as a transmission curve in the upper graph of figure 13. The logarithm of the thickness of the absorbing layer is plotted against the frequency cm -1. The various corresponding relative layer thicknesses are also shown in mm. In the photograph the absorption maximum occurs between 2,813 and 2,933 angstroms. It should be remembered that this is a transmission type curve. The numbers shown as frequency are wave numbers,
|
|
_1_λ |
|
|
v mm 1, where v = |
λ |
cm=cm 1. |
For the morphine curve, the maximum absorption occurs at 3,500 mm -1. The wave-length calculated to correspond to this frequency is
|
λcm= |
1 |
=0.00002857 cm |
|
|
35000 |
|
λ=2857 x 10 -8 cm=2857 ?=285.7 mμ
where 1 ?=10 -8cm. Hartley (1885) had determined the maximum wave-length to be 290 mμ according to the illustration in figure 9. Modern photometric determinations show a maximum at 285 m μ.
Kitasato classified the alkaloids into two major classes with the following basic structures ([38] ):
The following opium alkaloids were in class I, group III: cryptopine, protopine; group IV, narcotine, narceine; group V, papaverine, N-methyl-papaverine hydrochloride; group VI, cotarnine; the following alkaloids were in class II, group I, apomorphine; group II, thebaine; group III, morphine, codeine. The formulae used by Kitasato to represent thebaine, codeine and morphine, are as follows:
Thebaine Codeine Morphine
It is on this basis that Kitasato classified the alkaloids into the isoquinoline series. Thebaine according to Kitasato showed absorption at a "frequency" of 3,510 cm-1(wave number) or 284.9 mμ and morphine and codeine were shown as having identical absorption curves and a maximum at 3,485 mm-1 (286.9). The hypsochromic absorption of the two latter alkaloids in comparison with thebaine is explained by the lesser number of double bonds and the auxochromic effect of the methoxyl and hydroxyl respectively. The great difference between the light absorption of thebaine compared with morphine or codeine and compared with apomorphine was noted by Kitasato and attributed to the difference in the type of nitrogen ring. Apomorphine ultraviolet spectra obtained by different Hilger instruments were shown to have fine structure between 300 and 325 mμ as found by Gompel and Henri (1912), and Elvidge (1940) ([39] ). Recent determinations with the Beckman DU failed to detect the three small bands and showed a plateau in this region. Kitasato postulated that three ultraviolet characteristics could be explained since apomorphine has a pyridine ring connected to an aromatic nucleus while morphine has a pentamethylene-like structure connected to a hydrogenated ring as shown in the formula. The Gulland-Robinson structure is now generally accepted as the correct one for the series, and Kitasato's explanation has to be modified, in the light of modern theory.
Kitasato's study in the tetrahydroisoquinoline series showed that molecules containing a methylenedioxy or methoxy group have spectra with a single band at 3,200 ? for compounds with the auxochrome in the 1, 2 position and at 3,650 ? for compounds substituted in the 10, 11 position. Absence of 1, 2 substituents results in a flat curve at 3,200 ? while 9, 10 substitution instead of 10, 11 substitution has a bathochromic effect on the second band. Changes from tervalent (-N <) nitrogen to quinquivalent (R 1R 2R 3-NH)+ nitrogen were observed to exert hyper and bathochromic effects in the two classes with the structures III and IV.
O. J. Walker (1939) reviewed the literature on absorption spectrophotometry for the period from 1932 to 1938 and provided a comprehensive list of references from a large number of scientific journals covering this period ([40] ). The work on alkaloids and particularly narcotics was limited to 14 papers covering only alkaloids of the quinoline, ergot, strychnine, cinchona and berberine groups. Analytical, pharmacological and toxicological applications were described by V. Brustier (1932) and H. Fischer (1933) ([41] ).
Elvidge (1940) investigated the possibility of application of spectrometric absorption methods in the routine control of pharmaceutical preparations such as tablets and ampoules of a number of alkaloidal drugs for which chemical methods of assay were not generally available or applicable ([39] ). Absorption curves were given for the following narcotic and related alkaloids: morphine, diamorphine, codeine, codeine phosphate, and apomorphine hydrochloride; these were obtained in isopropyl alcohol and 0.5 N/HCl except for codeine. Morphine alkaloid was considerably altered in N/1 NaOH, a new absorption maximum occurred at 256 m μin place of the usual inflexion (246 m μ) which appears in the salts and freebase in alcohols, and a bathochromic shift of the λ max 276m μto 296 m μ. These data may be of use in distinguishing morphine from other non-phenolic alkaloids of the same general structure. Apomorphine showed a curve characteristic from the rest of this series, and had pronounced absorption at 273m μtogether with fine structure from 300 to 325 m μ. This fine structure, Elvidge stated, would well serve as a means of identifying small quantities of this alkaloid. Recent work has been unable to confirm presence of the fine structure.
Cocaine was studied by Ellinger in 1937 and its absorption spectrum recorded in the Tabulae Biologicae Periodicae (vol. XVI) ([42] ). Elvidge (1940) studied the spectra of cocaine and related cocaine substitutes as follows: hydrochlorides of cocaine, orthocaine, benzamine, amydricaine, phenocaine, amylocaine, procaine. The following four related structures give absorption curves of very similar shape: cocaine, amydricaine, amylocaine, benzamine. Water was used as a solvent in all cases, except with orthocaine, and in some cases the differences obtained by using water and alcohol as sol-vents were noted. The extinction values were expressed as
|
|
1% |
|
E |
|
|
|
1 cm. |
which expression is used when molecular weight is unknown. The influence of pH and solvent on shape of the ultraviolet absorption curves of these drugs is noted ([39] ).
Csoskan (1942) published an account of spectra of twenty opium alkaloids and derivatives ([43] ). He used a Zeiss chemical spectroscope, a Wolfram arc source, and a Winters silver grating; and photographed the comparison line spectra. The alkaloids for this work were obtained from S. Boehringer (Ingelheim-am-Rhein) and from E. Merck (Darmstadt). Double-distilled water and 96 per cent ethanol were used as solvents to make solutions of 0.001 molar concentration. The wave-lengths were recorded in millimicrons and log E values were plotted against wave-length to obtain the extinction curves.
Results showing maxima and log E values from Csoskan's work are tabulated in table III to compare with our findings. The curves in his publication were too small for practical use. Csoskan states that on account of the incompleteness of the experimental measurements the early work of Kitasato and Steiner can only serve for gross qualitative comparisons and that their maximum wave-lengths are in general agreement with his. In discussing the spectral curves of the opium alkaloids Csos- kan grouped the opium alkaloids into two classes according to molecular skeleton as follows:
I. Alkaloids with phenanthrene-isoquinoline skeletons
A. 1.Morphine, 2 Codeine, 3 Thebaine, 4. Neopine.
B. 1. Apomorphine, 2. Morphothebaine, 3. Epiapomorphine.
II. Alkaloids with the benzylisoquinoline skeletons
A. 1. Papaverine, 2. Narcotine, 3. Narceine, 4. Cotarnine and hydro-cotarnine, 5. Laudanine, 6. Laudanosine, 7. Cryptopine, 8 Protopine, 9. Gnoscopine.
Csoskan points out that the spectra of the Class IA alkaloids are very similar. The spectra of the apomorphine group are slightly different, morphothebaine being very different and having 3 band maxima. The alkaloids with a benzylisoquinoline skeleton are quite dissimilar, and the hydrastinin spectrum is similar to that of narcotine.
Salomon and Bina (1946) studied the ultraviolet spectrum of mescaline sulphate, the active component of the peyote cactus. These authors found an absorption range from 240-280 m μwith fine structure from 272-286 mμ ([44] ).
Strait, Kumler, Sah, Alpen and Chang (1948) discussed the ultraviolet absorption spectra of isomeric amidones and their structure ([45] ). They pointed out that the spectra are characterized by expected phenyl absorptions near 260 mμ and by anomolous absorbances near 300 mμ, and an unexpected strong band below 260 mμ in the spectrum of amidone and its nitrile in basic and hexane solutions in contrast with isoamidone and its nitrile. The differences (interchange of a methyl group between adjacent atoms) in the "accepted" structures of amidone and isoamidone and their nitriles are incompatible with these spectrum differences. They proposed that amidone has only one phenyl rather than the two "accepted" phenyl groups on the number four carbon atom to explain the spectrum characteristics. The spectrum character of isoamidone is compatible with the two "accepted" phenyl groups on the same carbon atom. They attributed the strong band below 260 mμ to the enol and enamine structures, thus possible only in amidone and its nitrile. The band at 290-300 mμ present in the amidones is associated with the α-phenyl carbonyl structure (Schultz, Robb and Sprague, 1947) ([46] ).
|
Band maxima | |||||||
|---|---|---|---|---|---|---|---|
|
Substance |
Solvent |
log E |
λmμ |
log E |
λmμ |
log E |
πmγ |
|
Phenanthrene |
|
|
|
|
|
|
|
|
morphine HCl |
H 2O |
- |
- |
3.08 | 282 | 3.49 | 242 |
|
morphine HCI |
C 2H 5OH |
- |
- |
3.11 | 287 | 3.50 | 242 |
|
codeine HC1 |
H 2O |
- |
- |
3.08 | 282 | 3.49 | 242 |
|
thebaine HC1 |
H 2O |
- |
- |
3.24 | 283 | 3.68 | 230 |
|
apomorphine HCl |
H 2O |
2.83 | 310 | 3.48 | 275 | 3.26 | 236 |
|
apomorphine HCl |
- |
- |
- |
3.42 | 264 | 3.33 | 224 |
|
morphothebaine HC1 |
- |
2.30 | 295 | 3.47 | 263 | 3.47 | 218 |
|
papaverine HCl |
- |
3.79 | 311 | 3.80 | 280 | 3.24 | 247 |
|
papaverine HC1 |
(Steiner) |
3.82 | 305 | 3.82 | 284 | 4.76 | 238 |
|
veratrol |
- |
- |
- |
3.35 | 275 | 3.86 | 226 |
|
papaverine HCl |
(synthetic) |
3.86 | 332 | 3.94 | 284 | 4.76 | 234 |
|
papaverine |
- |
3.43 | 325 | 3.50 | 274 | 5.54 | 237 |
|
papaverine |
(synthetic) |
3.43 | 325 | 3.76 | 272 | 4.58 | 234 |
|
narcotine HC1 |
H 2O |
- |
- |
3.46 | 310 | 3.21 | 280 |
|
narcotine HC1 |
C 2H 5OH |
- |
- |
3.61 | 299 | 3.40 | 280 |
|
hydrastine HC1 |
H 2O |
- |
- |
3.62 | 290 | 3.46 | 278 |
|
narceine HC1 |
H 2O |
2.07 | 375 | 3.75 | 285 | 3.73 | 231 |
|
cotarnine HCl |
H 2O |
3.96 | 333 | 3.93 | 252 | 3.96 | 212 |
|
hydrocotarnine HCl |
- |
- |
- |
3.14 | 286 | 4.10 | 215 |
|
benzylethylamine |
- |
- |
- |
3.30 | 279 | 4.21 | 246 |
Morgan (1949) and co-workers at the New York State Racing Commission Laboratory have applied ultraviolet spectrophotometry to a very large extent both qualitatively and quantitatively to determine many drugs extracted from urine and saliva samples from horses (47). The use of buffered drug solutions for measurement of ultraviolet spectra at different pH's has been made since at least 1949. They have utilized buffers instead of addition of acids. The buffers which are employed by Morgan et al. are listed in table IV.
St. John (1949) at Eli Lilly and Co. Laboratory in Indianapolis reported that a number of alkaloids inclu- ding morphine were being analysed by ultraviolet absorption spectrophotometry ([48] ).
Hubach and Jones (1950) described the use of ultraviolet spectra along with other optical properties, and microchemical reactions for identification of amidone hydrochloride. They listed maxima at 294 mμ, 259 mμ, and 292 mμ, having the following extinction coefficients: 460, 480, and 520.
Montesinos Ampuero (1951) studied the spectrophotometric determination of cocaine in solution by means of a Beckman spectrophotometer ([50] ).
Foster and MacDonald (1951) described the ultraviolet spectral changes of papaverine at different pH's. They discovered that the position of the max. of the absorption band was very pH dependent (51). Papaverine hydrochloride behaves as a very sensitive indicator in the ultraviolet.
Shaw and Jefferies (1951) determined the spectrum of phenadoxone,
|
|
1% |
|
|
λmax 259, E |
|
= 13.6; 292, 14.2 |
|
|
1 cm |
|
in water and in alcohol at 260 mμ, 12.0, at 294.5, 11.8 using a "Uvspek" photometer, concentration 0.1 per cent w/v, 0.5 cm cell ([52] ).
Stuckey (1952) reviewed the pharmaceutical analytical applications of ultraviolet absorption spectrophotometry from the period 1936 to 1952. In dealing with alkaloids, especially narcotics, mention is made of the work of Elvidge ( loc. cit.) on cocaine alkaloids and of the effect of pH on the morphine alkaloids.
|
Wave-length (millimicrons) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
pH |
Amounts of M/5 solu-tions of buffer constituents in 100 ml. of buffer |
220 |
230 |
240 |
250 |
260 |
270 |
280 |
290 |
300 |
310 |
| 1.1 |
HCl, 47.3 ml.
KCl, 2.72 ml. |
.011 |
.007 |
.004 |
.003 |
.003 |
.002 |
.001 |
.001 |
.002 |
.001 |
| 2.0 |
HCl, 5.95 ml.
KCl, 44.1 ml. |
.006 |
-.003 |
-.005 |
-.002 |
-.002 |
-.003 |
-.003 |
-.003 |
-.004 |
-.003 |
| 2.8 |
KH 2PO 4, 25.0 ml.
HCl, 3.50 ml. |
.004 |
.003 |
.003 |
.003 |
.005 |
.001 |
.002 |
.001 |
-.001 |
-.001 |
| 4.0 |
KH 2PO 4, 25.0 ml.
HCl, 0.25 ml. |
.004 |
.004 |
.001 |
.001 |
.002 |
.002 |
.001 |
.001 |
.000 |
.000 |
| 5.1 |
KH 2PO 4, 25.0 ml.
NaOH, 0.25 ml. |
.027 |
.024 |
.023 |
.023 |
.022 |
.015 |
.016 |
.014 |
.011 |
.010 |
| 5.8 |
KH 2PO 4, 25.0 ml.
NaOH, 2.82 ml. |
.039 |
.029 |
.021 |
.016 |
.015 |
.016 |
.015 |
.011 |
.010 |
.006 |
| 7.0 |
KH 2PO 4, 25.0 ml.
NaOH, 14.8 ml. |
.042 |
.030 |
.021 |
.018 |
.017 |
.015 |
.014 |
.011 |
.007 |
.004 |
|
7.9a |
H 3BO 3-KCl, 25.0 ml.
NaOH, 2.00 ml. |
.076 |
.050 |
.032 |
.026 |
.023 |
.023 |
.023 |
.018 |
.013 |
.009 |
|
9.0a |
H 3BO 3-KCl, 25.0 ml.
NaOH, 10.7 ml. |
.147 |
.105 |
.077 |
.067 |
.063 |
.058 |
.053 |
.045 |
.036 |
.030 |
|
10.0a |
H 3BO 3-KCl, 25.0 ml.
NaOH, 22.0 ml. |
.128 |
.063 |
.044 |
.036 |
.031 |
.025 |
.023 |
.021 |
.018 |
.022 |
| 10.8 |
Na 2HPO 4, 25.0 ml.
NaOH, 4.13 ml. |
.209 |
.101 |
.074 |
.065 |
.057 |
.053 |
.050 |
.045 |
.038 |
.032 |
| 11.8 |
Na 2HPO 4, 25.0 ml.
NaOH, 21.6 ml. |
.545 |
.163 |
.104 |
.089 |
.080 |
.070 |
.061 |
.052 |
.045 |
.038 |
These solutions are M/5 with respect to each constituent.
Stuckey recommended that conditions of analysis prevail which will ensure complete ionization and for solutions of weak bases a strongly acid pH is necessary to "stabilize" the spectrum so that it is unaffected by small pH changes. Stuckey states that the spectra of morphine, codeine and diamorphine (diacetylmorphine) as salts are very similar the absorption in acidic solutions being
|
|
1% |
|
|
low ( E |
|
; 280 m μca. 50); morphine in alkaline |
|
|
1 cm. |
|
|
|
1% |
|
|
solution shows a much higher absorption ( E |
; 258 m μca. 310, | |
|
|
1 cm. |
|
presumably due to the phenolic group. Apomorphine absorbs more strongly in acid solution
|
|
1% |
|
|
( E |
|
373 m μca. 600) |
|
|
1 cm. |
|
and this property can be used for its analysis in tablets and pharmaceuticals ([53] ).
Seagers, Neuss and Mader (1952) measured the ultraviolet spectrum of N-allylnormorphine in water solution and found a maximum at 285 m μand a minimum at 260 m μ([54] ). Addition of acid caused no change in the spectrum, but alkali caused a considerable increase in absorbance and a bathochromic shift of maximum to 298 and minimum shift to about 278 m μ. The ultraviolet chromophore present in N-allylnormorphine, morphine, normorphine, and related compounds, is the hexadrophenanthrene ring, according to Seagers et al., to which chromophore the authors attribute the similarity of the ultraviolet spectra of all opiates ([54] ).
Biggs (1952) has applied spectrophotometry to the detection and estimation of morphine in Stas-Otto extracts from viscera and natural products ([55] ).
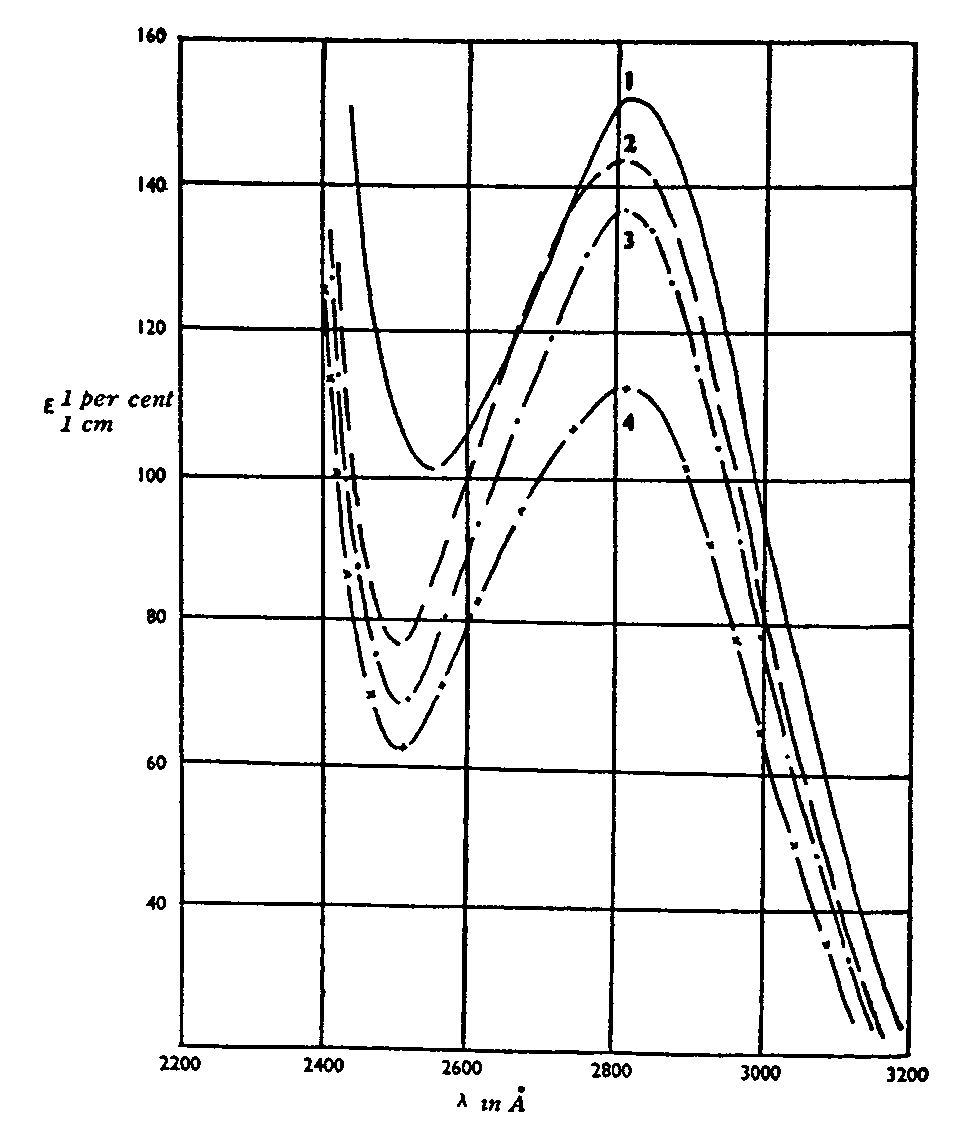
Spectra of Cannabis sativa extract, according to Biggs (1953)
Biggs (1953) examined the spectra of cannabis extracts and found their absorption curves are characterized by a band with pronounced ultraviolet absorption. Figure 14 shows extracts of Cannabis sativa, both saponified, and unsaponified, and cannabis extract from the viscera; figure 15 shows extracts of Cannabis sativa mixed with tobacco. The absorption bands are attributed to cannabinol and cannabidiol ([55] ).
Smith and MacDougall (1953) published a compilation of ultraviolet absorption data of compounds of toxicological interest ([56] ). This work has been critically evaluated by Morgan ([57] ). Brackett and Bradford (1953) have published a compilation of about 85 spectra of which 15 are narcotics ([58] ).
Clark and McBay (1954) have utilized the spectrophotometric spectra of morphine in different media at different pH's to determine these substances ([59] ). As means for identification of morphine and codeine the authors state: "This method of identification is valid only if the substance to be identified is suspected to be either morphine or codeine. Many substances, particularly other alkaloids of opium, may possess the same maxima as do morphine and codeine in alkaline media" (Baggesgaard-Rasmussen ([60] )).
Umberger (1954) in an excellent chapter on "Analytic Toxicology" describes the application and recent advances in the use of ultraviolet spectrophotometry for identification of drugs ([61] ). Kaye and Goldbaum (1954) in a recent text ([62] ) describe the application of spectrophotometry to the analysis of 33 drugs at two different pH values.
>For the greater part the review of literature has been up until now concerned with qualitative identification of narcotics by spectrophotometry. Quantitative determinations require a more precise knowledge of the influence of different variables upon the absorption energy. The simplest case of absorption is that of a parallel beam of monochromatic radiation passing rectilinearly through a homogeneous absorbing medium. The intensity of this beam is reduced by the same fractional part by each succeeding portion of its path under these conditions. Thus if the intensity is reduced by one-third in the first millimeter, it will be reduced to one-ninth of its original intensity by the second millimeter. This fact was first enunciated by Bouguer in 1729. It was later applied by Lambert, and sometimes is called Lambert's Law. According to Timma ([63] ) when dealing with solutions in which molecules are acting as energy absorbers, the optical path may be varied either by changing the cell thickness (see Hartley's early work, 1884-1907) or by changing the concentration of the solute. The latter method is often called Beer's Law. However, in a critical examination of Beer's paper (1852) ([64] ) Pfeiffer and Liebhafsky (1951) point out that ([1] ) Beer's paper does not contain an absorption law in which concentration appears as an explicit variable; ([2] ) Beer thought primarily of amount or mass of absorbing material; ([3] ) Beer may or may not have understood the functional relationship between absorbence and mass (or concentration) of absorbing material, at any rate he used length as an intermediate variable in correlating results obtained at different dilutions.
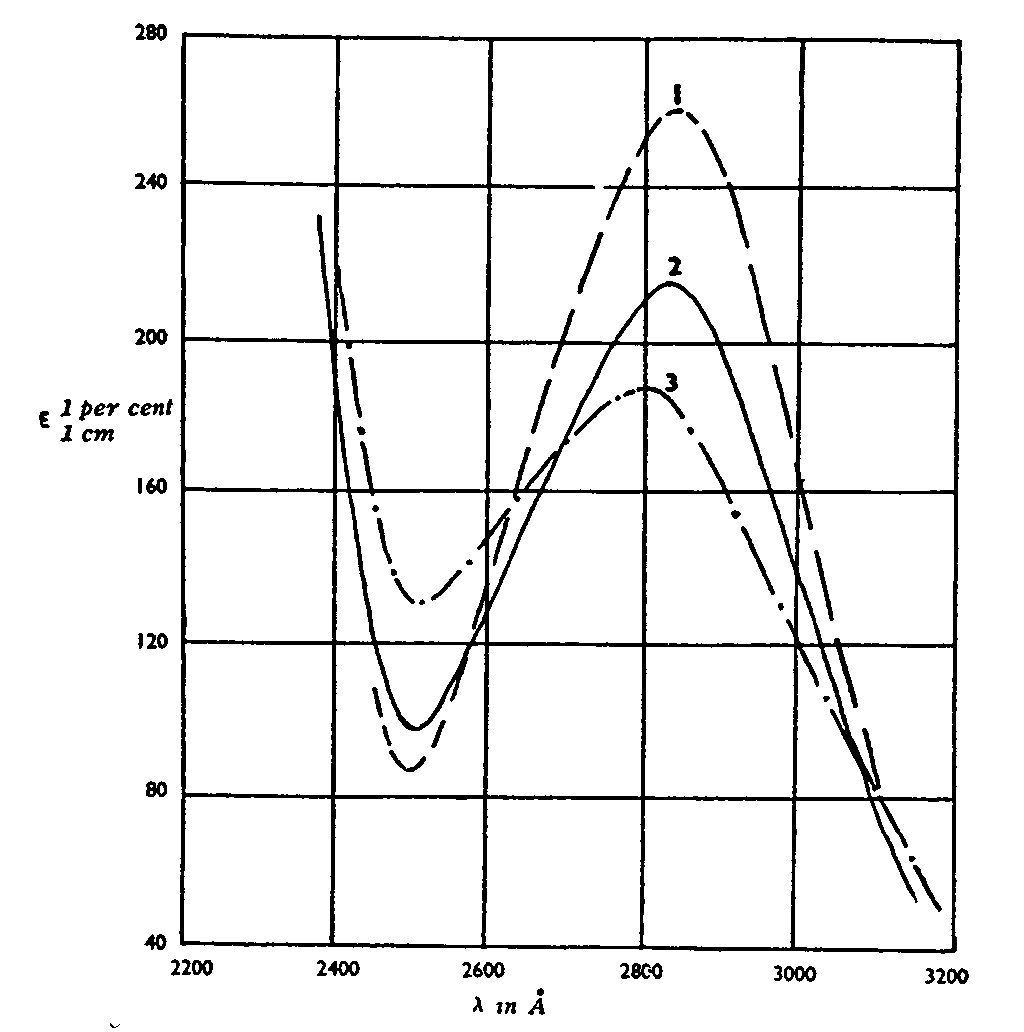
Spectra of Cannabis sativa mixed with tobacco, according to Biggs (1953)
There is no evidence in Beer's paper to indicate that he believed he had discovered a new absorption law. The phrase "Beer's Law" occurs forty years later (1889) in the writings of Walter. It was in Kayser's Handbuch (1905) that we find the statement of the attributing of the law to Beer as follows: "If we let a represent the absorption coefficient for unit concentration, there follows the absorption law
I=I 0 a d e
where d is the thickness of the layer and c the concentration. The law was set up by Beer and is called "Beer's Law". According to Pfeiffer and Leibhafsky: "There is thus good precedent for any twentieth century inaccuracies in describing what Beer thought and did."
Regardless of the origin the familiar expression of the Beer's Law is E=Kcd. where K = specific absorption coefficient, c is the concentration, and d is the sample thickness. Quantitative determinations consist of measuring the absorbency of a sample at wave-length or a narrow range of wave-lengths and correlating the extinction with concentration. Quantitative determinations involve the solving of simple algebraic equations (Friedel and Orchin) if the absorbencies are additive. If not additive quantitative determinations become empirical and depend upon the previous determination of a working curve showing the absorbency-concentration relationship.
While many instances are known where Beer's Law is said to be valid, deviations are both frequent and significant. Kortum (1936) has stated "that the validity of Beer's Law for ions must be considered as an exception rather than as a rule". Validity over large concentration ranges is not to be expected since Beer's Law is a limit law for low concentrations ([65] ).
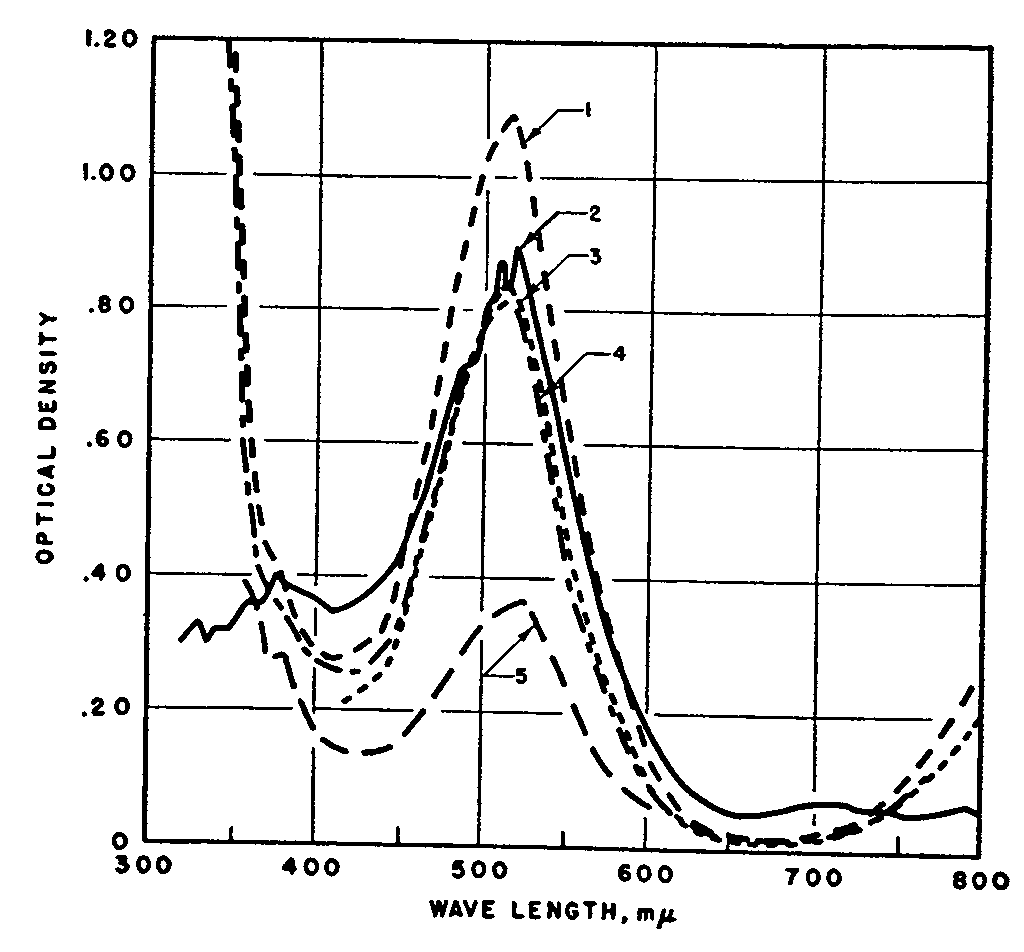
Absorption spectra of red colored solution produced from porphyroxine-meconidine by different spectrophotometers. Curve 1. Slit width 1 mμ at 512 mμ. Curve 2. Slit width 32 mμ at 512 mμ. Curve 3 . Slit width 1 mμ at 512 mμ and cell 1.3 cm Curve 5 . Lower concentration of colored material
The deviations from Beer's Law are of two types - instrumental and chemical or physical. It is generally held that a linear Beer's Law plot of absorbence
| log |
I 0 |
|
|
I |
versus concentration of an absorber is indicative of a linear response of the detecting system of the spectrophotometer. The results reported by Cannon and Butterworth (1953) demonstrate theoretically and practically that this is a fallacious assumption ([66] ). A nonlinear spectrophotometer, although giving a linear Beer's Law plot, will give erroneous values for absorptivity; absorbences are too low, i.e., the slope of the analytical calibration curve will be changed. Absolute tests of linearity of response of spectrophotometers should be made to determine the magnitude of the non linearity error in absorbence value. Other instrumental sources of error appear to have been restricted to the effects of stray light and finite slit width. What may be attributed to a deviation from Beer's Law may be a low degree of monochromaticity of the radiant energy used. Even the best monochromators give a spectral band of finite width, and apparent deviations will be noted if the absorbency of the sample changes considerably across the spectral interval isolated by the monochromator ([67] ) (Brattain, Rasmussen, and Cravath (1943), see figure 16). Poor matching of absorption cells and stray radiation may also be factors.
Physico-chemical changes in the absorbing material more often cause the deviations from the law than do failures in the law itself. Kortum and Seiler (1939) summarized the causes for divergence from the Beer's Law ([65] ). According to the theory of light dispersion it is not the specific absorption coefficient but the quantity Kn/(n 2+2) 2which is a constant, independent of concentration. For concentrations lower than 0.01 molar the change in refractive index, n, would rarely cause an error greater than the accuracy of measurements. Temperature fluctuation may influence absorption data in two ways. It may cause concentration changes due to expansion of the volume of solution, or it may cause a shift in the equilibrium constant of the absorbing system. Temperature effects may vary with different material. It amounts to 0.1 per cent per degree at 436 mμ for chromate ion and 1 per cent per degree for 2,4-dinitrophenolate ion. Equilibria may also be displaced by the presence of non-absorbing materials in the solution through the mass action law.
Many organic materials have weakly acidic or basic properties and their equilibria are easily disturbed by changes in acidity. For this reason it is often necessary to control the pH of the solution within narrow limits to obtain reproducible results.
The change in absorbence, location of wave-length of absorption maxima, and number of maxima of absorption spectra of many substances at different pH's has long been known. Increasing use of this property has been made both in qualitative and quantitative spectrophotometry.
About 1830 Stokes ([24] ) noted the increased fluorescence of quinine dissolved in sulfuric acid (see figure 17). Fischer ([33] ) (1925) related the enhanced ultraviolet absorbence of quinine in N/10 HCl and/or N/10 H 2SO 4to that of quinine sulphate in water alone. He showed the difference in the position and intensity of the absorption bands in acid and basic media. This work was done in the Forensic Medicine Institute of the University of Zurich.
Effect of pH on quinine sulfate, according to H. Fischer (1925)
In 1921 Pringsheim ([68] ) showed that aniline and naphthylamine solutions have absorption spectra which in hydrochloric acid become very similar to benzene and naphthaline absorption curves, similarly the quinine curve in hydrochloric acid becomes more like the quinoline curve. Brode (1924) showed that the change of pH value of solutions of indicators results in a change in intensity rather than a wave-length shift of the absorption band; thus one resonating system is eliminated and another is formed ([69] ). This change from one system to another is inversely proportional to the relative extinction coefficients, and hence there will be a point of extinction on the curves where the pH has the same value. Such a point is known as the isobestic point.
The effect of pH on the band maxima location of amines has been noted by many other workers. Kato and Someno (1937), Wohl (1939), Sklar (1939) showed that aniline has about 1/50 the absorption in acid that it has in water. Dede and Rosenberg (1934) explained this effect of resonance theory whereby the "lone" electrons on the nitrogen atom were unable to migrate from the amino group to the benzene ring in acid and thus the anilinium ion spectrum reverts to the benzene spectrum. Kumler and Strait ([70] ) extended this theory to fit any aromatic amino compound converted to a salt.
Kumler and Strait obtained a spectrum of sulfanilamide in sodium hydroxide with increased absorbances compared to the spectra in neutral and acid solutions. Bell, Bone and Rollin ([71] ) (1944) were unable to confirm this result and showed differences in spectra obtained by Kumler and Strait ([70] ) were due to absorption by sodium hydroxide solution. The importance of using ultraviolet transparent buffers in pH studies or else compensating blanks containing similar concentrations of ions should be noted. A set of ultraviolet transparent buffers for pH studies in this region was devised by Morgan ( loc. cit.) and is reproduced in table IV.
Vandenbelt and Doub (1944) made an extensive study of the absorption spectra of sulfanilamide derivatives and characterized them with respect to change in pH ([72] ). They were able to associate the spectral bands with absorbing groups in the molecule as follows: 257-259 mμ due to sulfanilamide portion, 280-283 mμ due to thiazole portion (258 mμ=thiazole structure) ; peak at 311 due to pyridine in sulfapyridine, band at 261 due to sulfanilyl portion, 241 mμ due to pyrimidine ring.
Stimson and Reuter (1946) ([73] ) studied the problem of estimating the methoxyl cinchona alkaloids in crude extracts. The four common natural occurring alkaloids and some related compounds were also measured over a pH range of 1-10. The maximum at 280 mμ in quinine, quinidine, epiquinine, and epiquinidine disappears and becomes a minimum at pH 4, 3, 1, and 1 respectively. There is a well defined invariant point (isobestic point) in the levo bases quinine and cinchonidine located at 295 mμ in both cases, which may be related to the optical configurations of these compounds and may show that the energy distributions accountable for the appearance of these isobestic points are destroyed by epimerism at either C 8 or C 9 of the molecule, as shown:
Morgan and Caprini (1947) made a further study of the effect of pH on sulfanilamide spectra to clear up the discrepancies in previous findings, and as a preliminary study for extensive work on the pH effect on spectra of many other drugs. A summary of the findings of various workers is included in table V ([34] ).
|
Acid or pH value |
λmax |
E |
Reference |
|---|---|---|---|
|
2 N HCl |
260 | 600 |
Vandenbelt and Doub |
|
0.8 N HCl |
260 | 620 |
Kumler and Strait |
|
pH 1.1 |
259 | 4,320 |
Morgan |
|
pH 7.0 |
258-259 |
16,160 |
Morgan |
|
M/1 NaCl |
258.5 | 16,380 |
Bell, Bone, Rollin |
|
M/1 NaCl |
258.0 | 14,130 |
Kumler and Strait |
|
pH 7.0 |
260 | 16,500 |
Vandenbelt and Doub |
|
pH 11.8 |
251.0 | 16,170 |
Morgan |
|
M/1 NaOH |
250.5 | 16,150 |
Bell, Bone, Rollin |
|
1N NaOH |
257.0 | 15,850 |
Vandenbelt and Doub |
|
Maxima at |
Minima at | ||||
|---|---|---|---|---|---|
|
Substance |
Solvent |
mμ |
E 1 per cent1 cm |
mμ |
E 1 per cent1 cm |
|
Morphine alkaloid |
Isopropyl alcohol and N/2 HCl |
282 | 56 | 263 | 19 |
|
Morphine alkaloid |
N/1 sodium hydroxide |
276 | 95 | 278 | 55 |
|
|
|
258 | 310 |
245-255 |
260 |
|
Diamorphine alkaloid |
Isopropyl alcohol and N/2 hydrochloric acid |
282 | 56 | 256 | 19 |
|
Codeine alkaloid |
Isopropyl alcohol |
279 | 42 | 265 | 20 |
|
Codeine phosphate |
Water |
280 | 44 | 265 | 30 |
|
Apomorphine hydrochloride |
Water and N/2 hydro-chloric acid |
273 | 670 | 248 | 220 |
|
|
|
305 | 200 | 299 | 130 |
|
|
|
315 | 165 | 310 | 145 |
|
|
|
324 | 120 | 321 | 115 |
In regard to the effect of pH on the spectra of phenols, naphthols and enols, Morton (1925), Tipping (1927) and Stenstrom (1925, 1926) showed that a shift to longer wave-lengths occurs when these hydroxyl compounds pass from neutral to basic solution since the ion absorbs at lower wave-lengths than the unionized compound ([74] ).
The effect of pH on the spectra of acids (salicyclic) and esters (acetylsalicyclic) was thoroughly studied by Edwards (1950). Below pH 1 each of these acids was regarded as undissociated, while above 6 the dissociation was substantially complete. The key points of the absorption curves are given in the table VIII ([75] ).
In 1940 Elvidge had determined the pH dependence of morphine, diacetylmorphine, codeine and apomorphine ([39] ). The spectral data in acid, base, and neutral (H 2O) solutions as determined by Elvidge are given in table VI.
Kumler et al. (1948) studied the effect of pH on the spectra of amidone etc. (see above). Foster and MacDonald (1951) studied papaverine spectra at a number of pH values. The results of their work are listed in table VII ([51] ).
|
pH |
Maxima E 1 %1 cm |
Minima at mμ = E 1 %1 cm | ||
|---|---|---|---|---|
| 1.5 | 250 | 1800 | 270 | 180 |
|
|
309 | 240 |
- |
- |
| 3.95 | 251 | 1595 |
- |
- |
|
|
- |
- |
- |
- |
| 6.3 | 245 | 1285 |
- |
- |
|
|
- |
- |
- |
- |
| 9.0 | 238 | 1700 |
- |
- |
|
|
- |
- |
- |
- |
| 11.0 | 238 | 1900 | 262 | 260 |
|
|
276 | 290 |
- |
- |
|
Compound |
Form |
λ(mμ) |
E |
|
|---|---|---|---|---|
|
|
Molecular |
236 | 8350 |
Maxima |
|
|
|
302 | 3620 |
|
|
|
|
261.5 | 300 |
Minimum |
|
|
|
228.5 | 6920 |
Maxima |
|
Salicylic acid |
Ionic |
296.5 | 3520 |
|
| 263.5 | 590 |
Minimum |
||
|
|
|
229.5 | 6890 |
|
|
|
|
249.0 | 1900 |
Isobestic |
|
|
|
298.5 | 3470 |
points |
|
|
|
327 | 510 |
|
|
|
Molecular |
275 | 1160 |
Maximum |
|
Aspirin |
|
259 | 760 |
Minimum |
|
|
Ionic |
None |
|
|
|
|
|
No isobestic points |
|
|
The carbinol-ammonium equilibrium is affected by pH and the spectra of cotarnine at different pH's reflect these shifts. This was first noted by Dobbie (1903). Hartley (1885), Dobbie (1903), Brustier (1926) and Kitasato (1927) were the first to measure the spectrum of narcotine and to illustrate the effect of pH on its spectrum data. These shifts are due to the change from a lactone form in acid to an alkali salt in base.
As a result of the increased interest in recent years, since about 1950, of scientists in the spectra of compounds at different pH's, several collections of spectral data for drugs at different pH's have been made. Morgan ( loc. cit.) has made an extensive collection most of which is unpublished. Smith and MacDougal (1950) have published ([56] ) a collection of data at two pH's for 154 different compounds. Goldbaum (1954) has recently published a list of about 85 compounds and spectral data in 0.5 N NaOH and 0.5 NHCl. Levi and Beaudry (1953) have collected the spectra of twenty-four barbiturates at 5 different pH's 3, 5, 9, 11 and 13. Brackett and Bradford (1952) have published 132 spectra. Beckman Instruments, Inc. (Nov. 1954) has offered to inaugurate a collection of ultraviolet curves of 50 common compounds at 3 pH levels. H. J. Noebels ([76] ) is in charge of this project. There is no doubt that these collections of spectra will be of potential value to analysts in the identification of drugs; however, cross-checking and comparison of the data in these above collections with one another and with data already in the literature from other sources indicates an alarming conflict. The maxima and minima appear to be in reasonable agreements but the extinction coefficients are very different, a factor of 8 times being noted in some cases. Morgan (1954) has already pointed out the necessity for critical review of the data before it is put at the disposal of forensic scientists. Compounds having spectra greatly influenced by pH should be thoroughly checked, others easily oxidized in alkaline solution have spectra which change rapidly with time. Impurities in some of the "standards" used should be checked, authenticity should be established as thoroughly as possible. Nomenclature of the compounds can be quite confusing and generic or chemical names should be used as often as possible in compilations. It should be possible for the scientists concerned to decide on a uniform method of presentation of data and a critical examination of the present material in the literature. Apparently forensic scientists are not alone in this state of confusion.
Qualitative data are generally presented as curves showing the absorbence as a function of the wave-length through the spectral range covered. No single method of presentation of these data has become universally accepted. In fact, Mellon ([77] ) (1949) has said "the confusion in usage, terms, symbols, and data in absorptiometry remains unsurpassed by that in any other division of quantitative analysis". One might add that there has been very little improvement in the past five years.
The ordinate is usually plotted in increasing values of transmittance, T, absorbency, A (also known as optical density or extinction) εor E, extinction coefficient, k, or absorbency index, as, molecular extintion, ε(or molar absorbency index, Am) and lately absorbence, or the logarithm of any of them. The abscissa is used to present the region of the spectrum investigated and may be expressed in one or more of the following terms: wave-length, λ, (in millimicrons m μ, or angstroms, ?); frequency, ν(in fresnels); wave-number, v or ν(in waves per centimetre); or the logarithm of the wave-length. Wave-length. wave-number and frequency are related by the expression
| 1 |
|
Frequency |
|
-------------- |
= Wave-number = |
-------------- |
|
Wave-length |
|
Speed of light |
With all the possible combinations in use including inconsistency in the direction of plotting, and disregard for general good graphing practice the literature is less than clear. Friedel and Orchin ([21] ) have gone so far as to present a scheme for transforming the various relationships with which one ordinarily deals ([2] ). Instrument measurements such as optical density (absorbence) and transmittance are often plotted directly from the instruments because of convenience, sometimes without any supplementary information regarding solvent, concentration, pH, etc.
Quantitative absorption spectrophotometry depends upon the correlation of the amount of energy absorbed at a given frequency with the amount of material present in the sample. This correlation is known as a working curve or calibration curve and consists of plotting the absorbence, or transmittance, as a function of concentration. Ringhom (1939) suggested plotting working curves of absorbency vs. the logarithm of concentration to emphasize the importance of making measurements within certain absorbence ranges where the error of measurement is smallest. If the transmittancy can be read to ±0.5 per cent, the maximum accuracy will be 1.4 per cent according to Peterson (1949). This is attained when the absorbency equals 0.4343 (36.8 per cent transmittancy). Ayres ([34] ) stated that the mini- mum error in absorbency occurs at 0.5550 (28 per cent transmittancy). Sobel ([34] ) has given the following table IX relating the concentration to absorbency, which was obtained from Morgan and Peterson's paper ([34] ).
Morgan ([34] ) advocates the use of the "baseline" procedure for calculating percentage in unknown solutions where the background absorption is considerable.
Absorption spectrophotometry offers a rapid accurate method for qualitative and quantitative analysis of narcotics. The historical development of the theory and application of these methods in relation to narcotics has been reviewed. The first work was done in 1885, and the first application in forensic cases was advocated at this time. About 1922, the use of spectrophotometry in the medico-legal field developed to a considerable ex- tent in Europe. The latest development in this field has been the use of the pH shift for characterizing spectra and thereby closely related compounds can be more easily discriminated.
|
Error in concentration, per cent | |||
|---|---|---|---|
|
Absorbency |
Transmittancy |
Scale ± 0.5% |
Scale ± 0.1% |
| 1.000 | 10 |
±2.17 |
±0.43 |
| 0.699 | 20 |
±1.55 |
±0.31 |
| 0.523 | 30 |
±1.37 |
±0.27 |
| 0.4343 | 36.8 |
±1.36 |
±0.27 |
| 0.398 | 40 |
±1.38 |
±0.28 |
| 0.301 | 50 |
±1.44 |
±0.29 |
| 0.222 | 60 |
±1.63 |
±0.33 |
| 0.155 | 70 |
±1.94 |
±0.39 |
| 0.097 | 80 |
±2.80 |
±0.56 |
| 0.046 | 90 |
±4.90 |
±0.90 |
BARTLET, J. C. and FARMILO, C. G., "The Determination of Countries of Origin of Opium Samples by Means of the Composition of the Opium Ash", United Nations document ST/SOA/SER.K/30, 12 July 1954.
002OESTREICHER, P. M., FARMILO, C. G., and LEVI, L., "Part III B: Ultraviolet Spectral Data for Ninety Narcotics and Related Compounds", Bulletin on Narcotics , vol. VI, No. 3.
003Beckman Division, Beckman Instruments Inc., South Pasadena, California. Booklet entitled: "What every executive should know about spectrophotometry".
004ALLEN, A. J., and FRANKLIN, R. G., "A Hydrogen Arc of High Intensity for Continuous Ultraviolet Radiation ", Journal of the Optical Society of America, 31, No. 3, 268-270 (1941).
005CARY, H. H., and BECKMAN, A. O., "A Quartz Photoelectric Spectrophotometer", Ibid., 31, No. 11, 682-689 (1941).
006Corning Glassworks, Corning, New York, Booklet: Glass Color Filters, 1946.
007Eastman Kodak Company, Rochester 3, New York, Wratten Light Filters, 1938.
008Farrand Optical Company, Bronx Blvd. and East 238th Street, New York 66, N. Y., Farrand Interference Filters, 1947.
009Baird Associates, Cambridge, Mass., U.S.A.- Interference Filters, 1946.
010HILGER, A. and WATTS, E. R., Hilger Division, 98 St. Pancras Way, Camden Road, London, N.W.I., Spekker Photometer (1937), H 237 and H 427.
011LOTHIAN, G. F., Absorption Spectrophotometry, Hilger & Watts, 1949. Available from Jarrell-Ash, U.S.A. Fig. 2 is a composite photograph taken from this text book with permission of the publishers.
012Figures 4 and 5 were drawings taken from the Tintometer publication, Electric Eyes, by A. J. FAWCETT, by permission of the Tintometer Ltd., Salisbury, England. (1951). This is a concise and elementary description of the photoelectric cell for the non-technical reader: its uses in industry, and its uses and shortcomings for colorimetry. The booklet contains a useful glossary of terms.
013KIRK, P., Quantitative Ultramicro Analysis, John Wiley & Sons, Inc., 440 Fourth Avenue, New York 16, N. Y. The section entitled "The Beckman Spectrophotometer" was taken for the most part from Kirk's text.
014HARRISON, G. R., LORD, R. C. and LOOFBOUROW, Practical Spectroscopy, Prentice Hall, New York. Table II.3. Chromophoric Groups, p. 275.
015International Critical Tables, vol. I. Literature references given for some common narcotics. Covering data to 1926.
016HENRI, V., and BRUNINGHAUS, L., Tables annuelles de constantes et données numériques, données numériques de spectroscopie, volumes I to XII.
017ROTH, W. A. and SCHEEL, K. A., Landold-Bornstein-Physikalische Chemische Tabellen. 5te ungearb. u. verm. aufl., von Walther R. Roth, u. Karl Scheel, Berlin, Springer 1923.
018BEILSTEIN, F. K., Handbuch der Organischen Chemie, 4th edition, Friedrich Richter, Berlin, Springer 1928.
019GMELXN, L., Handbuch der Inorganischen Chemie, 8th edition, Meyer, R. G. Leipzig, Verlag., Chemie, 1924.
020HIRT, R. C., American Cyanamid Co., Stamford, Connecticut. "Sources of Information on Ultraviolet Absorption Spectrophotometry". Reprinted from Journal of Chemical Education, vol. 29, 301-303, June 1952.
021FRIEDEL, R. A., and ORCHIN, M., Ultraviolet Spectra of Aromatic Compounds, John Wiley & Sons, Inc., New York. 1951.
022BREWSTER, D., "On the decomposition and dispersion of light within solid and fluid bodies", Edinburgh. Transactions 12 (1883), Ibid. 16 (1846), Phil Mag . (1848).
023MILLER, W. A., "On the Photographic Transparency of various Bodies, and on the Photographic Effects of Metallic and other Spectra obtained by Means of the Electric Spark", Journal of the Chemical Society, 17, 59 (1864).
024STOXES, G. G., "On the Change of Refrangibility of Light", Philosophical Transactions of the Royal Society, 107, Part II, 463 (1852): "On the long spectrum of electric light", Ibid., 152, II, 599 (1862): "On the application of the optical properties of bodies to the detection and discrimination of organic substances", Journal of the Chemical Society, 17, 304 (1864): "On a certain reaction of quinine", Ibid., 7 (1869): "On the discrimination of organic bodies by their optical properties", Phil Mag., 27, 388 (1864): "On the reduction and oxidation of the coloring matter of blood", Proceedings, Royal Society, London, (1864) Papers, vol. IV. 264 (1904)
025HARTLEY, W. N., "The Absorption Spectra of Alkaloids", Philosophical Transactions of the Royal Society, London, 176, II, 471-478, 516-517 (1885), Plates 54, 55 and 56.
026For a complete list of Hartley's works, see bibliography to Fischer, Hans, reference ([33] ).
027DOBBIE, J. J., LAUDER, A., and TINKLER, C. K., "The constitution of Cotarnine", Journal of the Chemical Society, London, 83, 598 (1903).
DOBBIE, J. J., and LAUDER, A., "On the relation between the absorption spectra and the chemical structure of corydaline, berberine, and other alkaloids", Ibid., 83, 605 (1903).
---------------- "The absorption spectra of laudanine and laudano-sine in relation to their constitution", Ibid., 83, 626 (1903).
DOBBIE, J. J., LAUDER, A., and TINKLER, C. K., "The relative strengths of the alkaline hydroxides and of ammonia as measured by their action on Cotarnine", Ibid., 85, 121 (1904).
DOBBIE, J. J., LAUDER, A., "The abstorption spectra of cinchonine, quinine and their isomerides", Ibid., 99, 1253 (1911).
---------------- "The absorption spectra of neopine", Ibid., 99, 34 (1911).
DOBBIE, J. J., and FOX, J. J., "The relation between the absorption spectra and the constitution of certain isoquinoline alkaloids and the alkaloids of Ipecaceuanha", Ibid., 105, 1639-42 (1914).
028HENRI, V., "Methode der quantitativen Messung der Absorption im Ultravioletten", Physikalische Zeitschrift, Vierzehnter Jahrgang, 1913, 515-516.
BIELECKI, J., and HENRI, V., Ibid., 516-521 (1913). See especially plate XVI facing page 514.
GOMPEL, M., and HENRI, V., "Absorption des rayons ultraviolets par les alcaloides du groupe de la morphine et par le phénanthrène", Comptes rendus hebdomadaires des séances de l'Académie des Sciences, 157, 1422-1425 (1913).
---------------- "Absorption des rayons ultraviolets par l'atropine, l'apoatropine et la cocaïne", Ibid., 74, 1066 (1913). Sec. Biologique.
029Approximately 30 papers were written by Henri, a complete list of which is given in Fischer's bibliography. See reference (33).
030HENRI, V., and ZANGGER, H., "Ueber Spektroscopie, Spectrographie und deren Anwendung", V. Naturf. Ges., Zurich, 65, 334 (1920).
031ZANGGER, H., "L'évolution des méthodes de spectroscopie, de spectrographie et leurs applications en médecine légale", Annales médecine légale, 2, 145 (1922).
032STEINER, P., "Les spectres ultraviolets de la narcotine et de ses produits de décomposition", Annales médecine légale, 2, 338 (1922).
FISCHER, H., and STEINER, P., "Les spectres d'absorption ultraviolets de la pyridine et de l'isoquinoléine", Compte rendu, etc. 175, 882 (1922).
STEINER, P., "Les spectres d'absorption ultraviolets des alcaloides du groupe de l'isoquinoléine. La narcotine, l'hydrastine et l'hydrocotarnine", Ibid 176, 244-6 (1923); "Les spectres d'absorption ultraviolets du veratrol et de la vanil-line", Ibid., 176, 744 (1923); "Les spectres d'absorption ultraviolets des alcaloides du groupe de l'isoquinoléine. La papavérine et son chlorhydrate", Ibid., 175, 1146-9 (1922); "La narceine", Ibid., 176, 1379-81 (1923); "Etude spectrographique des alcaloides végétaux. L'absorption des rayons ultraviolets par les alcaloides de classe de l'isoquinoléine et de la morphine", Bulletin de la Société chimique biologique, 6, 231 (1924).
033FISCHER, H., "Die Anwendung der Spektroskopie in der Gerichtlichen Medizin als Feststellung Methodik speziellzum Toxikologischen Nachweis von Alkaloiden. Experimentellen Teil: Die Absorptionspektra der Chiningruppe und der sie aufwendenden Substanzen", Inaugural Dissertation zur Erlangung der Doctorwürde der medizinischen Fakulat der Universität, Zurich, vorgelegt von Hans Fischer, Pract. arzt. von Schaffhausen, Assistenz Arzt am gerichtlich medizinischen Institut der Univ. Genehmigt auf Antrag von Herrn Prof. Dr. H. Zangger, Zurich, 1925. Buchdruckerei Stafa, A. G. Vorna E. Gull. aus dem Institut für physikalische chemie der Universität, Zurich. Direk. Prof. Dr. v. V. Henri. This book contains a very complete bibliography on spectroscopy covering the years up to 1925.
034MORGAN, C. E., Chief Chemist, Division of the State Racing Commission, Department of State of New York, 148-11 Hillside Avenue, Jamaica 35, New York.
035BONTEMPI, L. A., "Absorption spectra of morphine solutions in the ultraviolet" (translated title, C.A.), Anales soc. cient. argentina, 99, 209-228 (1925).
036EISENBRAND, J., "Recent progress in the absorption spectra analysis of dissolved substances" (translated title), Pharm. Ztg., 71, 716 (1926).
037BRUSTIER, V., "Contributions ? l'étude spectrographique de l'absorption des rayons ultraviolets par les alcaloides et les glucosides", Bull. Soc. Chim., 39, 1527-1543 (1926); "Ultraviolet absorption spectra of alkaloids", Chim. et Ind., 27, 1007 (1932).
038KITASATO, Z., "Beitärge zur Kenntnis der Isochinolin Alkaloid'', Acta Phytochimica (published at Tokyo by the Iwata Institute of Plant Biochemistry), vol. II, No. 2, 175-257 (1927).
039ELVIDGE, W. F, "Absorption spectrophotometry in pharmaceutical analysis" I & II, Quarterly Journal of Pharmacy & Pharmacology, 13, 219-36 (1940).
040WALKER, O. J., "Absorption spectrophotometry and its application'', Bibliography and Abstracts, 1932 to 1938. Published by Adam Hilger, 98 St. Pancras Way, London, N.W.I. England, 866 references.
041FISCHER, H., "Study of cocaine and other alkaloids of toxicological importance", Archives, exptl. Path. Pharmakol., 170, 610 (1933).
042ELLINGER, F., "Absorption Spektroscopie in Ultraviolet", Tabulae Biologicae Periodicae, Band VI, 1937, II, Band XVI, pars. 4, (1938).
043CSOSKAN, P., "The absorption spectrum of opium alkaloids", Magyar Chem. Folyóirat, 47, 7-20 (1941): "Spectroscopic detection of opium alkaloids", Z. anal. chem., 124, 344-50 (1942).
044SALOMON, Kurt and BINA, Albert F., "Ultraviolet absorption spectra of mescaline sulfate and β-phenylethylamine sulfate", Journal of the American Chemical Society, 68, 2403 (1946).
045STRAIT, L. A., KUMLER, W. D., SAH, P. P. T., ALPEN, E. L., and CHANG, F. N. H., "The ultraviolet absorption spectra and the structures of Isomeric Amidones", Proceedings of the Optical Society of America, Journal of the Optical Society of America, 38, No. 12, 1098 (1948).
046SCHULTZ, E., ROBB, G., and SPRAGUE, J., "The reaction of 1-diamethylamino-2-chloropropane with diphenylacetonitrile. The structure of amidone", Journal of the American Chemical Society, 69, 2454 (1947).
047MORGAN, C. E., private communications may be obtained by writing the author. See reference (34).
048ST. JOHN, C. V., "Spectrophotometric determination of alkaloids and other compounds absorbing ultraviolet radiation. I. Determination of Quinine". Bulletin of the National Formulary Committee, vol. XVII, Nos. 11 & 12, Nov. & Dec., 208-213 (1949).
049HUBACH, C., and JONES, F. F., "Methadone Hydrochloride Optical Properties, Microchemical Reactions & X-ray Diffraction Data", Analytical Chemistry , 22, 595 (1950).
050AMPUERO-MONTESLN0S, F., "Absorpción ultravioleta de la cocaina", Rev. Facultad farm. y bioquím., 13, Nos. 49/50, 43-63 (1951).
051FOSTER, G. E., and MACDONALD, J., "The ultraviolet spectrum of papaverine hydrochloride", Journal of Pharmacy and Pharmacology, 3, 127-128 (1951).
052SHAW, W. H. C., and JEFFRIES, J.P., "The Determination of Phenadoxone", Ibid., 3, 823-827 (1951).
053STUKEY, R. E., "The applications of ultraviolet spectra, absorption spectrophotometry in pharmaceutical analysis", Ibid., 4, 345-365 (1952).
054SEAGERS, W. J., NEUSS, J. D., and NADER, W. J., "The Identification and Determination of N-allylnormorphine Hydrochloride", Journal of the American Pharmaceutical Association, Scientific Edition, XLI, 640-642 (1952).
055BIGGS, A. J., "The Spectrophotometric Identification and Estimation of Strychnine, Brucine and Morphine in Viscera Extracts", Journal of Pharmacy and Pharmacology, 4, 547-559 (1952), "The Spectrophotometric Detection of Cannabis Sativa Resin", Ibid, 5, 18-25 (1953).
056SMITH, H. W., and MACDOUGAL, J. R., "A Compilation of Spectrophotometric Data on Compounds of Toxicological Interest", Attorney General's Laboratory, Toronto, Canada, May 1953.
057MORGAN, C. E., private communication. See reference (34).
058BRACKETT, J. W., and BRADFORD, L. W., "Detection, Identification, and Estimation of Non-Volatile Organic Poisons, Drugs and Narcotics by Ultraviolet Spectrophotometry", Manuscript presented at 123rd meeting of the American Chemical Society, Los Angeles, California, March 1953.
059CLARK, W. A., and MCBAY, A. J., "Spectrophotometric Determination of Morphine and of Codeine", Journal of the Pharmaceutical Association, Scientific Edition, XLIII, No. 1, Jan., 39 (1954).
060BAGGESGAARD-RASMUSSEN, M. H., "Recherches sur la morphine". Extrait des Annales pharmaceutiques francaises, Nov.-Dec., Tome X, 693 (1952).
061GONZALES, I. A., VANCE, M., HELPERN, M., and UMBERGER, C. J., "Legal Medicine Pathology and Toxicology", Sections 41 to 47. "Analytic Toxicology by UMBERGER, C. J., 946 to 1298, Appleton-Century-Crofts Inc., New York, 1954 (second edition).
062GRADWOHL, R. B. H., "Legal Medicine", Chapter 24 "Toxicology" by Sidney Kay and Leo R. Goldbaum, 599-723, published by C. V. Mosby Co., St. Louis, Mo., 1954.
063TIMMA, D. L., "Absorption Spectrophotometry", The Ohio Journal of Science, 52 (3) 117, May, (1952).
064PFEIFFER, H. G., and LIEBHAFSKY, H. A., "The Origins of Beer's Law", Journal of Chemical Education, 28, 123-125 (1951).
065KORT?M, G., and von HALBAN, H., "Zur Methodik der relativen und absorbaten lichtelektrischen Extinktions Messung", Zeitschrift für Physikalische Chemie, A, 170, 212-230 (1934). "Das optische Verhalten geloster Ionen und seine Bedeutung fur die Struktur electrolytischer Losungen". V "Lichtabsorption and Dispersitat organischer Farbstoffionen in wässeriger Lösung", Zeitschrift für Physikalische Chemie, B. 34, 255-274 (1936), and Seiler, M. "Die kritische Auswahl colorimetrischer, spectralphotometrischer und spektrographischer Methoden zur Absorptionsmessung", Angewandte Chemie, 52, 687-693 (1939).
066CANNON, C. G., and BUTTERWORTH, I. S.C., "Beer's Law and Spectrophotometer Linearity", Analytical Chemistry, 25, 168 (1953).
067BRATTAIN, R. R., RASMUSSEN, R. S., and CRAVATH, A.M., "A Spectrophotometric Method for the Analysis of Multi-component Mixtures and its Infrared Applications", Journal of Applied Physics, 14, 418-428 (1943).
068PRINGSHEIM, P., "Fluoreszenz und Phosphoreszenz im Lichte der neuen Atom Theorien", J. Springer, Berlin, 1921, Seite 167.
069BRODE, W. R., "The Determination of Hydrogen-ion Concentration by a Spectrophotometric Method and the Absorption Spectra of Indicators", Journal of the American Chemical Society, 46, 581 (1924).
070KUMLER & STRAIT, "The ultraviolet absorption spectra and resonance in benzene derivatives-Sulfanilamide, metanilamide, p-aminobenzoic acid, benzenesulfonamide, benzoic acid and aniline", Ibid. 65, 2349 (1943). See this article for earlier references to Kato & Someno, Wohl, Sklar & Dede & Rosenberg.
071BELL, P. H., BONE, J. F., ROLLIN, R. O., "The relationship of structure to activity of sulfanilamide type compounds", Ibid. 66, 847 (1944).
072VANDENBELT, J. M., and DOUB, L, "The ultraviolet absorption spectra of simple unsaturated compounds. I. Mono and p-disubstituted benzene derivatives", Ibid., 69, 2714 (1947).
073STIMSON, M. M., and REUTER, M. A., "Spectrophotometric estimation of methoxycinchona alkaloids", Ibid., 68, 1192-1196 (1946).
074See Lothian for references to the work of MORTON (1925), TIPPING (1927) and STENSTROM (1925-26).
075EDWARDS, L. J., "The Hydrolysis of Asprin", A determination of the thermodynamic dissociation constant and a study of the reaction kinetics by ultraviolet spectrophotometry, Transactions of the Faraday Society, 46, 723 (1950).
076Newsletter, American Academy of Forensic Sciences, Nov. 1954, No. 16. Dr. Irving Sunshine, Cuyahoga County Coroner's Laboratory; 2121 Adelbert Road, Cleveland, 6, Ohio.
077MELLON, M. G., Analytical Absorption Spectroscopy, published by Wiley & Sons, New York (1950), p. 82.

