Natural narcotics and alkaloids are used therapeutically either in the form of natural vegetable drugs, of which they are the most effective component, as various galenic preparations made from these drugs, or in the form of chemical compounds isolated from these drugs or synthesized. In the last instance they occur most frequently in the form of salts readily soluble in water, or as suitable pharmaceutical preparations, the latter applying almost exclusively to synthetic narcotics. In evaluating narcotics and alkaloids the special conditions of the instances mentioned above are to be respected. Determining salts is relatively the simplest case. Many accepted methods depend on determining the acid component with the aid of titration (hydrochlorides by argentometry, sulphates by alkalimetry, etc.). This method of determination cannot be considered fully adequate, as it is indirect and does not determine the physiologically active component, i.e., the organic base; furthermore it does not take into account the not entirely stoichiometric neutralization of the base during the manufacture of such salts. Because of these reasons Van Os 1 prefers accepted methods which determine the organic base directly, as he notes in his criticism of the British Pharmacopoeia.
Author: Antonin Jindra
Pages: 20 to 26
Creation Date: 1955/01/01
Natural narcotics and alkaloids are used therapeutically either in the form of natural vegetable drugs, of which they are the most effective component, as various galenic preparations made from these drugs, or in the form of chemical compounds isolated from these drugs or synthesized. In the last instance they occur most frequently in the form of salts readily soluble in water, or as suitable pharmaceutical preparations, the latter applying almost exclusively to synthetic narcotics. In evaluating narcotics and alkaloids the special conditions of the instances mentioned above are to be respected. Determining salts is relatively the simplest case. Many accepted methods depend on determining the acid component with the aid of titration (hydrochlorides by argentometry, sulphates by alkalimetry, etc.). This method of determination cannot be considered fully adequate, as it is indirect and does not determine the physiologically active component, i.e., the organic base; furthermore it does not take into account the not entirely stoichiometric neutralization of the base during the manufacture of such salts. Because of these reasons Van Os [1] prefers accepted methods which determine the organic base directly, as he notes in his criticism of the British Pharmacopoeia.
These methods for determining the bases are, however, much more complicated and resemble those used for chemical evaluation of vegetable drugs and galenic or pharmaceutical preparations containing narcotics and alkaloids. These methods are always based on the disengaging of the organic base and its extraction or elusion by a suitable solvent. Very often these processes tend to be very cumbersome, require considerable amounts of solvents and substances to be analysed, and take a long time regardless of whether volumetric, gravimetric, or some other method has been chosen. When determining narcotics and alkaloids in vegetable drugs and galenic preparations it is, moreover, necessary to remove any inconvenient accompanying substances. It is often necessary to depend on determining the total contents of various chemically similar compounds, which cannot as yet be separated by any current method.
Because of the reasons stated there is an incessant trend to find simpler specific methods suitable for the determination of narcotics and alkaloids.
In the course of time all chemical and physico-chemical methods introduced into chemical pharmaceutical analysis have been put to this use. With a small number of these methods it has been possible to cover the entire broad field of narcotics, while the majority were suited to particular alkaloids or for closely related groups of them. In principle, these were titration methods with indicator or electrometric signalling of the end point of the titration, as well as colorimetric, spectrophotometric, polarographic and chromatographic techniques.
With visual neutralizing titration, suitably chosen indicators are indispensable. By determining electrometrically the concentration of hydrogen ions, or by electrometrical titration, the selection of the proper colour indicator has been greatly facilitated; this raised the accuracy of the visual determination. Müller,[2] Kranz,[3] as well as Masucci and Moffat[4] used hydrogen electrodes and found a great variety of hydrogen ion concentrations in non-purified alkaloid salts. This shows that the salt need not be formed by the two components in strict equivalence; one component may predominate in case of the manufacturing process being imperfect. The quinhydrone electrode has been used-among others-by Wagner and Gill [5] as well as by Rasmussen and Schou.[6] Kolthoff and Hartong [7] used the potentiometric method and an indicating antimony electrode for the titration of alkaloid salts. Later on Rasmussen and Reimers [8] determined the dissociation constants of alkaloids dissolved in ethanol by means of colorimetry and electrometry; they proposed suitable indicators for alkaloid titrations in water-ethanol mixtures as well. Saunders [9] and Srivastava give potentiometric titration curves for various alkaloid salts in water and water-ethanol media together with the pK b values of the respective alkaloids. Jindra and Pohorské [10] have found glass and antimony electrodes very suitable for the potentiometric titration of alkaloid bases. Domange [11] in a survey cites the potentiometry of a great number of various narcotics and alkaloids.
Conductometric titrations of alkaloids and their salts were carried out by Kolthoff.[12] Polarometric titrations have been used to good advantage in determining some narcotics. Jindra, Kalvoda and Záka [13] titrated morphine, apomorphine, and dilaudid salts by p-diazobenzensulfonic acid on the basis of a coupling reaction; Jindra, Jungr and Záka [14] used the same agent for cephaeline and emetine. Polarometric titration by silicotungstic acid may be used in the determination of nicotine.[15] Silver or lead nitrate with polarometric indication may be used for the titration of alkaloids and narcotic salts in mixtures by precipitating their anion components. Thus Kalvoda and Záka [l6] have determined the hydrochlorides and sulphates of morphine and atropine as well as a mixture of quinine sulphate and hydrochloride. Morphine is determined with the aid of polarimetry by oxidizing it by excess of potassium bichromate to oxydimorphine and establishing the quantity of remaining excess reagent by titration with a lead nitrate solution; the titration end-point is indicated visually or polarimetrically. The method mentioned [17] may be used to determine morphine in laudanum, and in opium and its extracts, in addition to the remaining alkaloids contained in these substances. Recently the possibility has been established of using solutions of substances current for the determination of alkaloids as volumetric agents for precipitation polarometric titrations. Záka [18] quotes a solution of iodine and potassium iodide, forming complex substances (e.g., potassium triiodomercurate, potassium triiodoplumbite, dichlorotetrahydroxoplantinic (IV) acid, gold trichloride), picric and picrolonic acids, styphnic acid, Reinecke's salt, silicomolybdic acid, silicotungstic acid. The titration of organic bases with the aid of a high frequency oscillometer has been accomplished by Wagner and Kauffman.[19]
Levi, Oestreicher and Farmilo [20] have treated the neutralizing titration of narcotics in non-aqueous media. The results of their research show that by means of titration it is possible to determine organic bases in non-aqueous media even in the form of salts; perchloric acid in acetic acid is used as the titration agent. Similar titrations have been reported by Riddick,[21] Pifer and Wollish,[22] Ekeblad,[23] and Pifer, Wollish and Schmall.[24]
In some instances the chemical structure of the organic base allows Volumetric methods other than neutralization to be used for the determination. Thus, e.g., in bromometry, a solution of bromine in glacial acetic acid is used as the volumetric agent and its excess is determined by iodometry. In this way many different alkaloids have been determined, especially morphine derivatives (Tomecek and Záka; Tomecek, Kucera and Záka.[25] There is a colorimetric method for the determination of nearly every narcotic; these methods make use of the colour change, occurring during the reaction of the respective narcotics with the various agents in suitably chosen conditions of medium temperature, concentration and time. Very often a great number of diverse methods are cited for one substance. Dealing with this question in greater detail would pass beyond the limits of this paper; the same applies to ultraviolet and infra-red spectrophotometry. Let it suffice to note in this respect Stucke's survey [26] on ultraviolet spectrophotometry.
Polarography is often a useful tool for determining the effective component of numerous narcotics. Reduction waves are the result of alkaloids, the carbonyl group of which is conjugated with double bonds, aromatic nucleus or an oxygen link or, finally, of alkaloids containing a quinoline or isoquinoline nucleus. Recently the polarographic determination of lobeline was accomplished (Uffelie[27] ), as well as that of veratrum alkaloids (Walaszek and Pircio [28] ), oxycodeinone, dihydromorphinone, etc. (Santavy,[29] Volke and Fortova [30] ). The Baggesgaard-Rasmussen nitrosation method found various applications (Matsumuto [31] ). Similarly it is possible after nitrosation to determine cephaeline, even in the presence of emetine (Jindra, Jungr., Zy ka[32] ). Even nicotine[33] and strychnine[34] are capable of being reduced; there is also a description of determining scopolamine.[35]
The chromatographic technique was found useful in analysing narcotics and alkaloids very soon after its renaissance twenty years ago. At first absorption chromatography, later partition chromatography, and finally chromatography on ion exchangers were applied.
Valentin and Franck [36] were the first to use this method for the easy determination of cantharidine in tinctures and later on Franck applied this method to evaluate medicines and galenic preparations yielding typical absorption zones on alumina (Merck puriss. anhydr.). Furthermore, Merz and Franck [37] have established that during transit of alkaloid drugs through an alumina column, not their coloured components only, but their anions are being captured as well. After elution by 70% ethanol, alkaloid bases could be found in the nearly colourless output liquid; these bases could be determined by titration after evaporating the ethanol. A method was developed to determine the alkaloid content in tinctures and extracts of Atropa Belladonna, Quina, Strychnos nux vomica, and Ipecacuanha. In the following years this work gave an impetus to other workers to exploit this promising technique.
First of all some narcotics have been separated; Kondo [38] separated morphine from thebaine, Levi and Castelli[39] used chromatography to separate morphine, codeine, narcotine, and papaverine. Björling[40] uses an analogous elusion method for the determination of Strychnos nux vomica tincture and extract. He also carried out a blank experiment in order to establish the alkalinity of the absorbent and checked up on the method experimentally with pure strychnine nitrate salt. Ljungberg[41] tried miscellaneous kinds of alumina with the object of determining liquid quina extract; for dissolving and elution he made use of 90% ethanol. Mühlemann and Tobler [42] simplified the determination of colchicine in galenic preparations by using an alumina column and developed a chromatographic titration method for the determination of alkaloids in Cola acuminata seeds and in galenic preparations made up of them. La Rocca and Burlage[43] succeeded in separating strychnine from brucine in preparations of Strychnos nux vomica seeds.
In the laboratory of the Danish Pharmacological Commission in Copenhagen, Reimer, Gottlieb and Christensen,[44] in the years 1943-45, searched for possibilities of determining alkaloid salts chromatographically. By direct titration of the basic component they determined the salts of cocaine, emetine, physostigmine, pilocarpine, scopolamine, and ethylmorphine, as well as salts of synthetic organic bases, i.e., procaine, percaine, pantocaine, and pethidine, using this unified method: approximately 0.2 grammes of the salt is dissolved in 5 millilitres of ethanol (86%). The eluate is diluted with an equal amount of water and titrated with 0.1 N solution of hydrochloric acid to a green colour with bromphenyl blue.
For various other salts it was necessary to modify the method suitably, either because the elution of the alkaloid was slow and imperfect or on account of the anion passing into the solution simultaneously. The decisive factors were the height of the chromatographic column, the absorption power of the alumina as well as the amount and concentration of the ethanol used in the process. The authors state precise conditions for the analysing of atropine, codeine, ephedrine, homatropine, hyoscyamine, quinine, quinidine, strychnine, and eucodal salts. Their technique cannot be applied to the analysis of salts of narcotine, papaverine, amphetamine, apomorphine, morphine, or of any of the salts of alkaloids with weak polyvalent acids, where the elution of the base is very slow. According to the authors' opinion the behaviour of the alkaloid base on the alumina column is determined by the anion character as well as by the basic component. Björling [45] too has found the elution of some organic bases and polyvalent acids to be slow and imperfect. According to his opinion the elution may be improved by adding acids, bases, or salts to the ethanol solution of the alkaloid salt.
The authors devoted much attention to the quality of the alumina used, as it showed different absorption power and basicity; and they describe a method of standardizing it. Correct concentration of the ethanol used is of importance too, because in a diluted ethanol medium the alumina partly releases hydroxyl and the analytical results are higher. On the other hand, too high absorptive power is a disadvantage too, as elution of the base is not complete and the results are too low. This method found very favourable reception by Van Os.[1] According to him the alumina must not be even weakly alkaline and must have satisfactory absorptive power; he also gives tests which the alumina must pass in order to render the chromatographic method reliable. The method is used by Ph. Dan. IX for the determination of salts of organic bases.
Brown, Kirch and Webster[46] analysed chromatographically drugs of the Solaneaceae genus, thus continuing Franck's work. They found alumina to be the best suited absorption material. They modified the USP method by absorption of ether extracts of alkaloid drugs and accelerated the process considerably without sacrificing accuracy and simplicity. Their results were greater than those reached according to the accepted method; experimentally they found out the reason, which was not the presence of other nitrogen bases but the ease of operation and the minimum losses of alkaloids. Their work was continued by Lasslo and Webster,[47] who determined the alkaloids in ipecacuanha roots. They used ammonia-ether extracts of the pulverized drugs, inspissated the solution, dissolved the remainder in sulphuric acid and after expelling the ether completely captured the alkaloids in a special instrument on a Forisil column. They consider Florisil the best material-as compared with alumina, calcium hydroxyde, lactose, etc. -to separate alkaloids from any accompanying substances; the exchanger material-Amberlite IR 100 and Zeo-Karb-proved unsuita ble as well because it dissolved in the alkaline elution agent and resulted in coloured solutions. The synthetic zeolite Decalso did not reach the absorptive power of Florisil. The elution of the base from the column was accomplished by 10% ammonia-ethanol; after evaporating the eluting agent the emetine was determined by titration.
Florisil has been used by Klee and Kirch[48] to determine morphine in opium as well. The method is as follows: extraction of the morphine from the opium by means of boiling methanol and elution from the Florisil column by the same solvent in the device as used by Stolman and Stewart[49] for absorption analysis of morphine, codeine, and heroin in biological material. The meconic acid and the coloured substances interfering with the colorimetric determination proper of the morphine by means of the Folin-Ciocault agent remain absorbed on the column.
Partition chromatography makes use of a great variety of materials. Guani and Ganguly[50] studied the capturing of many various alkaloids on silica gel. Evans and Partridge[51] used buffered silica for the separation of alkaloids from Solaneaceae plants. Munier and Macheb&oeliguf,[52] as well as Brindle, Carless and Woodhead[53] used chromatography on paper to separate a great variety of alkaloids. Romeike[54] succeeded in separating tropine alkaloids with the aid of planar diaphragm filters and applied this method to their quantitative analysis as well. She determined them colorimetrically after leaching the alkaloids out of detected stains. Morphine may be determined[55] in a similar way by leaching it out of the paper and using polarography after separation.
Ion exchangers have been used for experiments with alkaloids at a time when only artificial zeolites of mineral origin were available; Ungerer[22] followed the exchange of organic bases, including alkaloids, from their salts on calcium permutite. According to his findings the exchange in ethanol medium takes place in the sense of equivalence; by plotting the results he obtained typical Freundlich absorption isotherms. The free base precipitated from the aqueous alkaloid salt solutions under the influence of the weakly alkaline calcium permutite and its capturing grew apparently higher.
Oberst[57] absorbed morphine from urine extracts on permutite and followed the quantitative properties of this process in the case of aqueous as well as ethanol solutions. Fischer and Chalupa[58] used various absorbents to purify extracts of biological material in analysis of substances with narcotic effects. McIntosh, Kelsey and Geiling[59] captured morphine on permutite from 50% ethanol solutions during their work with C 14 morphine. The morphine is disengaged by a substance the ionization constant of which is greater than 7.4 x 10 -7, and is not eluted by petroleum ether, ether, carbon tetrachloride, acetone, or absolute methanol. The authors followed the capturing of morphine from other media (chloroform) as well as the capturing of other opium alkaloids (papaverine, narcotine, codeine and thebaine) from a great variety of solutions, and treated their elution.
Artificial and synthetic zeolites did not seem suitable for the analysis of alkaloids on account of their pores being too small for the exchange of the large alkaloidal cations. Recently, however, Björling and Berggren [60] turned to this type of material in their study of chromatographic alkaloid analysis; the reason was that resinous exchangers capture the alkaloids from the solutions well, but their quantitative elution is often difficult. They chose Decalso, a synthetic silicate of the permutite type acid. Iron must be removed from the material as far as possible, because it interferes with the spectrophotometric determination proper of the alkaloids, if hydrochloric acid in greater quantity is used for the elution. Each analysis requires a fresh absorbent. On the activated Decalso the alkaloids are quantitatively removed from the salt solutions and may be completely eluted by means of acids or mineral salts.
Their process is as follows: an aqueous solution of the alkaloid salt (0.7-5.0 milligrammes) is poured onto the column of activated Decalso (0.25-0.6 grammes) in a tube of 5 millimetres diameter. Water is used for the elution and this is followed by a sufficient quantity (4 millilitres approx.) of 0.02 N-HCl, 0.2 N-H 2SO 4 or of potassium (sodium) chloride in 25 % solution. The alkaloid contents in the eluate are determined by measuring the extinction in the maximum point of the absorption curve with the aid of a spectrophotometer.
The authors used this method at first for the determination of atropine sulphate and methylbromide as well as hyoscine hydrobromide and methylnitrate; they also elaborated an application for the analysis of solutions, suppositoria, tablets, and extracts, containing the substances mentioned. Later the authors used the same method to determine salts of morphine, codeine, papaverine, berberine, hydrastinine, neostigmine, d-tubocurarine, and oxedrine.
The discovery of sulfonated coals and of synthetic organic exchangers opened up new vistas; especially the preparation of resinous substances with exchanger characteristics by Adams and Holmes.[61] In the case of alkaloids of course there was the tendency to make use of these new substances for isolation purposes (Nachod,[62] Applezweig[22] ). During preliminary experiments the capturing intensity on quinine from a sulphuric acid solution was observed, using the sulfonated carbonaceous Zeo-Carb zeolite. Applezweig carried out the elution of the alkaloid base by ammonia-ethanol and thus combined the regeneration of the material with the elution of the quinine. Applezweig and Ronzone[64] used the same exchanger for industrial extraction of alkaloids from quina bark and Applezweig[65] used an analogous method to obtain atropine from Datura. Kingsbury, Mindler and Gilwood [66] described their method of obtaining nicotine by means of Zeo-Carb too. Later on the trend changes towards the exploitation of synthetic exchanger resins; these became commercially available in various grades of special characteristics and standard quality.The author of this survey has found out during preliminary experiments with alkaloid capturing on the cation exchanger Amberlite IR 100, that this exchanger is not suited for quanti- tative work with various alkaloid salts. Winter and Kunin [67] discovered during their work with the weak cation exchanger Amberlite IRC 50 of the carboxyl type, that alkaloids may be released from it by an almost stoichiometric quantity of acid. When capturing alkaloid salts it is necessary to transform the exchanger into a salt. Weak bases, however, are captured in small quantities only, thus enabling this exchanger to be used for separating weakly basic alkaloids (strychnine, caffeine) from stronger ones.
Saunders and Srivastava [68] followed the degree of absorption and the elution speed of quinine from 50 % ethanol solution on Amberlite IRC 50 in its acid form. They also used a resin column, from which they obtained quinine in various purely quantitative ways. They remark that the absorption capacity of the resin is raised considerably by very fine granulation. The exchange however proceeds very slowly and the equilibrium is reached only after several days. In another work the authors [69] studied the equilibrium state of various organic bases (ephedrine, nicotine, caffeine, quinine) using their aqueous and ethanol solutions and weak cation exchangers. The degree of absorption as well as the capacity of equilibria depend on the dissociation constants and on the size of the molecules of the substance to be absorbed. Huyck[70] followed the capture of ephedrine from an ethanol solution on Dowex 50 and on Amberlite IRC 50. According to Saunders[71] weak cation exchangers are useful for alkaloid determination, especially in case of the solution being coloured or polluted. The base is especially successfully absorbed from moderately alkaline ethanol solution. As soon as the alkaloid has been captured onto the exchanger, the column undergoes elution by a suitable solvent to remove the impurities and the alkaloid is removed by means of ammonia, if it is a weak base (quinine) or by means of hydrochloric acid, if the alkaloid is strongly basic (ephedrine). The determination proper is carried out according to a suitable technique.
Buchi and Furrer[72] presumed that in aqueous solutions alkaloid salts form cations suitable for exchange. With regard to the cation size special requirements are laid down for the exchanger. The authors tried some modern cation exchangers as far as their use for quinine hydrochloride is concerned. They tried to find the best form of resin, the most advantageous medium and the best elution technique for the captured quinine. It became apparent that Amberlite IR 120 and Dowex 50, being resins of the polystyrene type, are not well suited for the exchange of so large a cation. Amberlite IRC 50 showed very little exchange activity in acid and ethanol medium. The best resin found was of the sulfon type-Duolite C 10-on which quinine exchange was quantitative from strongly acid as well as ethanol solutions. Complete elution of the captured quinine was accomplished best by 10 % ammonia solution in ethanol. The method was applied to the determination of alkaloids in quina extracts and bark; its results-when compared with the Ph. Helv. V. method-were very successful.
With respect to its phenolic hydroxyl, morphine is of weakly acid character. Grant and Hilty[73] have shown that the morphine base is captured on the strongly basic anion exchanger Amberlite XE 75; this enables the separation of morphine from codeine, which is not captured on the column, but passes into the solution and may be titrated. The captured morphine may be obtained quantitatively from the column by elution with diluted acid and may be determined spectrophotometrically.
From these statements it appears that capturing alkaloid bases on modern exchangers, and even to a greater degree their disengaging and elution, are not always successful. Alkaloids do form large size cations capable of exchanging in aqueous solutions, but this exchange takes place in sufficient measure only in the case of pores in the resin mesh being of greater diameter than the ion diameter of the alkaloid cation. The large alkaloid cations; bound very strongly to the resin mesh by the attractive forces, may be disengaged by the methods current for inorganic ions with difficulties and imperfectly only. The elution must be carried out in some special way, e.g., according to Sussman, Mindler and Wood,[74] who disengage the alkaloid by a stronger base; the alkaloid base is not dissolved in water to any observable extent, so that it remains in the mesh and may be removed from there by the use of a suitable solvent. The use of an alkaline organic solvent is a welcome simplification. In determining alkaloid salts, the necessity of base elution from the column may be evaded by determining the acid component-after capturing the base on the column-with the aid of titration. This was the method chosen by Jampolskaja,[75] who determined arecoline bromide, eukodal, benzedrine, and carbocholine as well as other pharmaceutical preparations. The main disadvantage of this method is of course that it determines the non-physiological acid component only.
A different approach was chosen by the author of this survey after he had convinced himself that the cation exchanger Amberlite 100 was unsuitable for quantitative analysis of several current alkaloid salts. He adopted the following method: the acid components were captured on the strongest anion exchange then available-Amberlite IR-4b-and the disengaged alkaloid base was brought out of the column with the aid of elution by hot methanol or ethanol. The base contents in the colourless eluate were determined by titration, using a mixed indicator (methyl red and methylene blue). In this way the author determined the salts of strychnine, atropine, morphine, brucine, ephedrine, quinine and cinchonine. He described the preparation of the exchanger column and a method of its regeneration by a carbonate solution, thus enabling one column to be used many times. The choice of the solvent is important, because the salt as well as the alkaloid base must be readily soluble in it. The rate of flow is important in some instances only (atropine sulphate). The author pointed to the possibility of using this method for the analysis of galenic preparations and by doing so he opened up new perspectives for work in this field.
In the following works Jindra and Pohorské[76] focused their attention on the application of the chromatographic ion exchange method for the determination of all alkaloid salts of the Czechoslovak Pharmacopoeia (CSL 1) and for the elaboration of a standard semimicro- and micro-method. In this manner they determined: arecoline hydrobromide, atropine sulphate, cocaine hydrochloride, codeine phosphate, homatropine hydrobromide, morphine hydrochloride, papaverine hydrobromide, pilocarpine hydrobromide, quinine hydrobromide and sulphate, scopolamine hydrobromide, and strychnine nitrate. With some salts the method was not successful. Thus apomorphine decomposes oxydatively in ethanol solutions and has to be titrated in a nitrogen atmosphere, while physostigmine is also unstable and in its analysis lower results were found consistently. The same applies to ephedrine hydrochloride and cotarnine chloride. On account of the higher basicity of ephedrine its acid component is bound more strongly and the Amberlite IR-4b exchanger is not able to separate the two components quantitatively. An analogue to ephedrine is the salt of cotarnine, which is a strong quaternary base and is split only imperfectly by a weak anion exchanger. The authors have predicted the possibility of determining even these substances with the aid of a stronger anion exchanger. Their prediction was confirmed by the work with Amberlite IRS 400, in the course of which Jindra and Rentz[77] determined ephedrine hydrochloride in an analogous manner.
The method elaborated for the determination of alkaloid salts is very fast, economical, accurate and advantageous, as it determines the physiologically active component. The authors also devoted their attention to the determination of the contents of vegetable drugs and galenic preparations containing alkaloids. The substances in question were : Quina bark, Ipecacuanha root, the seed of Strychnos nux vomica, Atropa belladonna herbs, Datura herbs, and their preparations. In the majority of cases it was necessary to isolate the alkaloid to some degree before the chromatographic process proper could begin and in many cases the authors simplified the isolation process considerably as compared with the accepted methods. As small quantities of samples and solvents were used, the rate of work was made considerably faster. When galenic preparations were chromatographed directly without previous removal of the accompanying substances, the results obtained were often much higher. This may be explained by the presence of amines in the drug; these amines are subject to titration together with the alkaloids. In addition there is the splitting of base or ammonia salts, which makes itself felt especially in the case of small sample quantities.
The authors' method has been generalized to determine the salts of other organic bases; according to Saunders[78] it is much simpler and easier than, for example, the method of distilling with water vapour in the case of ephedrine salts (Ph.B.1953) or the extraction methods of various pharmacopoeias. Thus Jindra and Rentz[79] used Amberlite IRA 400 to determine some narcotics and sympathomimetic amines in their salts and tablets; e.g., amphetamine, pervitine, ephedrine, privine, and ephetonal; all these are substances in which the benzene ring is not substituted by a hydroxyl. Such a substitution (adrenalin, sympathol) renders the determination of the substance in question impossible, because it makes the substance weakly acid and this results in its reaction with the exchanger being imperfect. Jindra and Rentz[80] determined the salts of some current anaesthetics: procaine, larocaine, tutocaine, percaine, amylocaine, amethocaine, and diocaine. Jindra and Motl[81] determined the contents of antihistaminics (antistine, n?oantergane, phenergane, probedryle, antihistamine) as well as other narcotics and organic bases (acedicon parpanit, syntropan, trasentin, atebrin[82] ,[83] ) in tablets and dragées according to the instructions quoted at the end of the survey.
Baggesgaard-Rasmussen, Fuchs and Lundberg[84] turned to synthetic exchangers as well, because they considered methods current in analysing alkaloids until now to be too cumbersome; the reason is the great number of operations to be performed as well as the insufficient reliability because of the chance of alkaloid losses during evaporation of the solvent. These authors used Amberlite IRA 400; they followed its solubility and proceeded in accordance with the principle and process outlined above. In order to obtain greater accuracy they did not use commercial samples of alkaloid salts and other organic bases, but free bases, the values of which they established by titration with bromphenol blue in 50 % methanol or ethanol solution. The elution was carried out by 50 % ethanol in most cases; exceptionally they used more concentrated ethanol (75 %) or methanol, or even hot solvents. A strongly basic exchanger was not suitable for morphine salt analysis; physostigmine decomposed; and histamine, carbacholine, and homatropine methylbromide yielded no satisfactory results. The authors found the Amberlite IR-4b exchanger unfit for atropine-salt analysis. According to their opinion, total conversion does not take place on this exchanger and the authors excluded the possibility of atropine absorption on the column. In this respect there is an antagonism in relation to the works of other authors, who obtained correct results with the same exchanger, using elution with 95 % hot ethanol or concentrated methanol.
Levi and Farmilo[85] found the ion exchangers to be suitable material for narcotics analysis. They proceeded according to the same principle as outlined above, using Amberlite IR-4b and ascertained that narcotics may be determined by this method with + 1.6 % accuracy; they praise the method as being simple, fast and using small sample quantities as compared with the accepted methods, where the organic base is determined by the classical extraction method. All the authors stress the importance of keeping the disengaged base in the solution; its secretion makes quantitative elution difficult. Thus - according to their opinion -the choice of a suitable solvent or mixture of solvents is of primary importance for a successful analysis. The authors used methanol, methanol-chloroform, methanol-ether, and ethanol-chloroform. They determined salts of methadone, phenadoxone, alphaprodine, cocaine, pethidine, codeine, diamorphine, dihydromorphine, morphine, and papaverine, and applied the method to the valuation of pharmaceutical preparations. In case of such analysis it is necessary to remove any inert insoluble material. No ionizing impurities or other substances may be present, which would take part in the exchange process in an undesirable way. The method proved its value in analysis of tablets containing salts of codeine, procaine, morphine, pethidine, and heroin.
Gundersen, Heiz and Klevstrand[86] consider synthetic exchangers to be more advantageous for chromatographic analysis of alkaloid salts than alumina, mainly because of the possibility of regenerating and using the same column over and over again. They determined alkaloid salts and other organic bases along the same lines as those quoted previously and focused their attention especially on the choice of a suitable exchanger. At present there is such a synthetic material available commercially; it is suited for the exchange of anions with analytic purity and graded power. The authors have chosen a powerful anion exchanger, Dowex 2, which they prefer to the analogous Amberlite IRA 400, as they consider it especially suitable for analysis requiring a resin in the form of a free base. On this exchanger the salts split quantitatively, and its regeneration is easy. To dissolve the salts and for the elution of the base, 70% ethanol was used; amphetamine required a larger quantity of solvent for elution, whereas the base of atropine needed 96 % ethanol for elution. The authors determined the salts of cocaine, codeine, ephedrine, hyoscine, lobeline, mepacrine, methadon, pethidine, procaine, and strychnine. They titrated the disengaged base with potentiometric checking or using bromphenol blue as an indicator.
With Amberlite IRA 400 it is not possible to analyse morphine and other bases of phenolic character in this manner, as these substances are held as anions. On the other hand, this fact may be used to separate morphine from other alkaloids; e.g., from ephedrine hydrochloride, as the authors have done too. Dowex 2 hydrolyzes some alkaloids; e.g., pilocarpine, yohimbine and neostigmine. The authors used the method to analyse substances containing salts of amphetamine, codeine, ephedrine and pethidine. They evaluate the method as more accurate, faster, and more suitable than the methods used heretofore.
From the facts mentioned it appears that ion exchangers have already found their place and application in pharmaceutical analysis, as there are commercially available grades of standard composition. The methods making use of them are incorporated in the Czechoslovak Pharmacopoeia (CsL 2) for the determination of salts of narcotics, alkaloids, and other organic bases. Additional experiences about the physico-chemical conditions during the exchange of organic ions on exchangers of various types surely will bring about further simplification of the old analytical processes of the substances proper as well as of their mixtures. Modern ion exchangers, however, have in pharmacy a scope of application much broader than for analysis only, as may be seen in recent surveys by Kressmann,[87] Saunders [78] and Jindra and Motl.[88]
Resinous ion exchangers are high molecular polymers made up of interlinked long carbon-atom chains so that a huge mesh is formed. The respective types of synthetic resins differ by the contents of acid or basic groups, which are mostly bound to benzene groups and render feasible the characteristic exchange qualities. The ion exchange characteristic remains intact even in its bonds within the resin, so that an exchanger with sulfon groups is more acid than an exchanger containing carboxyl groups. Similarly exchangers of the aromatic amine type are more weakly basic than those of the aliphatic amine or quaternary base type. Thus we distinguish among cation and anion exchangers as well as among strong and weak ones.
Modem exchangers are sold in the form of small balls. They are not soluble in water, acid and basic media, nor in organic solvents. According to the chromatographic technique columns of them may be prepared on which the exchange between the resin and the solution may proceed quantitatively. Each exchanger is characterized by its capacity, depending on the number and character of functional groups, degree of swelling, which is determined by the transversal chain linking, grain size, etc.
The reactions on the exchangers are of various types. A cation exchanger may operate in the hydrogen cycle or in the cycle of some salt. During the exchange between a cation exchanger of the sulfon type in the hydrogen cycle and the electrolyte solution (e.g., NaCl) the anions of the exchanger attract the cations of the metal, which permeates the resin mesh and the disengaged hydrogen ions may be brought out by elution using chromatographic technique.
(RSO 3-H +)+Na +Cl -<==>(RSO 3 -Na +)+H +Cl -
The reactions on anion exchangers are explained either by the absorption of acid or by anion exchange. Most probably both reactions are taking place to a certain extent. When determining alkaloid salts on the anion exchanger the following exchange may be considered:
(RNH 2/H 3O+OH -)+A.HX+H 2O<==> (RNH 2/H 3O/X -)+A+2H 2O
The used exchanger may be regenerated; in the case of cation exchange an excess of acid is used for this purpose, whereas after exchanging anions an excess of base serves for regeneration.
The state of equilibrium was studied with much care in the case of organic as well as inorganic ion exchange. Until now, however, only partial solutions were obtained, based on general schemes or empirical equations and combining the effects of both diffusion and electrostatic forces. The difference of interpreting the exchange may be explained by the type of exchanger used. In synthetic permutites the exchange was considered to be a purely chemical reaction in which the law of mass preservation prevails. Later on the exchange interpreted as an absorption phenomenon. Furthermore, there is the theory of the crystal lattice, according to which the attraction and repulsion of the ions is the result of electrostatic forces. The theory of the double layer explaining the electrokinetic properties of colloids was applied to the ion exchange as well. Most important for synthetic resinous ion exchangers is the Donnan theory of diaphragm equilibria, which in a special application explains various phenomena observed in ion exchange. Ion exchange is always governed by the laws of electro-neutrality and demands the presence of active exchange centres, which realize the combination of the resin with the ions carrying opposite charges.
When analysing salts of narcotics and alkaloids by the chromatographic method of ion exchange 67, 76, 84, 85, 86 it is necessary to arrange the chosen column and to proceed according to accurate instructions for use. The exchanger resin is pulverized in a porcelain mortar with a small amount of water until a grain diameter of 0.05-0.5 mm is reached; by a larger amount of water the grains are transferred to a beaker. The suspended fine resin fragments are then decanted from the settled coarser grains and after adding water the entire process is repeated; then the resin is left under water for at least 48 hours. During this time the water is decanted several times so that the resin is ready for the preparation of the column; in this form it may be kept under water in a closed container. Several authors recommend an elution of the exchanger by water and ethanol (95%) and transfer it to the OH cycle by hydroxide solution and run water through it until neutrality is realized. After drying, the substance is screened and grains of 0.34 mm average size are obtained.
The authors used chromatographic tubes of various diameters.
The prepared resin is dispersed in a small amount of water and at the same time the cock is opened. The resin with the water is poured in without interruption so as to prevent the column from sucking in air bubbles. Thus, the amount of resin required for the respective methods is introduced into the pipe and the column is consolidated by a cotton wool swab; then the exchanger is regenerated by hydroxide or carbonate, and water is passed through it until neutrality is reached. The effluent liquid must be quite colourless. The throughput rate is approximately 2-3 millilitres with the cock fully open. Thus the column is prepared for the experiment proper, after which it must be regenerated and washed out with water.
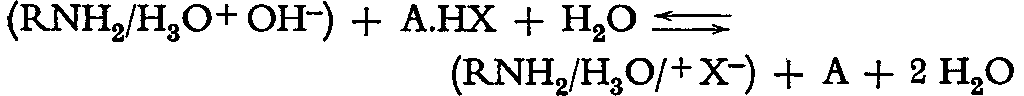
Instructions for carrying out the experiment:
Method according to CsL 2:
Approximately 0.1 gramme of the organic base is weighed accurately and dissolved in 10 millilitres of ethanol (80 %) in a 50-millilitre beaker. By opening the cock the water above the resin column (10 mm diameter, 10 cm high) is let out so that its level sinks to the cotton wool level. At this instant hot ethanol, total quantity 30 millilitres, is poured onto the column in doses of 5 millilitres each so that each successive quantity of the hot ethanol is added at the instant of its level almost reaching the level of the cotton wool. After the last dose the hot solution to be analysed is poured onto the column in two doses in the same manner without interrupting the flow through the column. The elution of the column and of the container is carried out in the same manner by a total quantity of 50 millilitres of hot ethanol (80 %). The eluate is filled into a titration flask (250 millilitres) beginning with the instant when the first dose of salt solution was poured and ending with the instant when the last dose of elution ethanol reached the cotton wool level; then it is diluted by 50 millilitres of water not containing carbon dioxide so that it is ready for the titration which is carried out by 0.1 N solution of hydrochloric acid; the end-point is determined by a mixed indicator, methyl red and methylene blue, or it may be potentiometric, with a calomel and antimony electrode.
Semimicromethod:
Twenty to thirty milligrammes of the alkaloid salt are dissolved in 10 millilitres of 80 % ethanol. The elution is carried out by 20 millilitres of hot ethanol (96 %). Approximately 5-8 grammes of exchanger are used. A blank experiment is carried out with the solvents used.
Micromethod:
Five milligrammes of alkaloid salt are dissolved in 5 millilitres of 80 % ethanol; a column of 1.5 grammes of exchanger in a chromatographic tube of adequate dimensions is washed out by 5 millilitres of hot ethanol and the warm alkaloid salt solution is chromatographed quantitatively. The elution is carried out by 10 millilitres of hot ethanol (96 %) in several doses. The titration is potentiometric using 0.01 N hydrochloric acid solution.
Determination in tablets and dragées:
One or two tablets or dragées containing 25-100 milligrammes of substance are leached in 10 millilitres of 50 % ethanol for a sufficient period of time (1-4 hours). The suspension thus obtained is filtered through cotton wool and the fillers are extracted several times by a total amount of 20 millilltres of boiling ethanol (96%). The combined filtrates are chromatographed on the exchanger, using the method described in the case of determining salts.
VAN Os, D., J. Pharm. Pharmacol., 1, 55, 350 (1949).
002MULIER, F., Z. Elektrochemie, 30, 587 (1924).
003KRANZ, J. C. Jr., J. Am. Pharm. Ass., 14, 294 (1925).
004MASUCCI, P., MOFFAT, M. I., J. Am. Pharm. Ass., 12, 609 (1923).
005WAGNER, L. R., GILL, W. J., J. Am. Pharm. Ass., 14, 288 (1925).
006BAGGESGAARD-RASMUSSEN, H., SCHOU SV. Aa., Z. Elektrochemie, 31, 189 (1925).
007KOLTHOFF, I. M., HARTONG, B. D., Rec. Trav. Chim., 44, 113 (1925).
008BAGGESGAARD-RASMUSSEN, H., REIMERS, F., Dansk Tidskr. Farm., 9, 235 (1935).
009SAUNDERS, L., SRIVASTAVA, R. S., J. Pharm. Pharmacol., 3, 78 (1951).
010JINDRA, A., POHORSKY, J., Cas. ces. l?k?rn., 63, 57 (1950).
011DOMANGE, L., J. Pharm. Pharmacol., 4, 513 (1952).
012KOLTHOFF I. M., Z. anorg. Allgem. Chem., 112, 196 (1920).
013JINDRA, A., KALVODA, R., ZYKA, J., Collection 15, 797 (1950).
014JINDRA, A., JUNGR, V., ZYKA, J., Cs. farm. 1, 186 (1952).
015DE ANGELIS, B., Ric. Sci. 21, 62 (1951).
016KALVODA, R ., ZYKA, J., Cas. ces. l?k?rn. 63, 36 (1950).
017BLAZEK, A., KALVODA, R ., ZYKA, J. Cas. ces. l?k?rn. 62, 69 (1949) KALVODA, R., ZYKA, J., Cas. ces. l?k?rn. 62, 134 (1949).
018ZYKA, J., unpublished data.
019WAGNER, W. F., KAUFFMAN, W. B., Anal. Chem. 25, 538 (1953).
020LEVI, L., OESTREICHER, P. M., FARMILO, Ch. G., Bull. on Narcotics V, 1 (1953).
021RIDDICK, J. R., Anal. Chem. 24, 41 (1952).
022PIFER, Ch. W., WOLLISH, E. G., Anal. Chem. 24, 300 (1952).
023EKEBLAD, P., J. Pharm. Pharmacol. 4, 636 (1952).
024PIFER, Ch. W., WOLLISH, E. G., SCHMAIL, M., J. Am. Pharm. Ass. 42, 509 (1953).
025TOMICEK, O., ZYKA, J., Cas. ces. l?k?rn. 62, 49 (1949). TOMICEK, O., KUCERA, R., ZYKA, J., Cs. farm. 1, 344 (1952).
026STUCKE, R. E., J. Pharm. Pharmacol. 4, 345 (1952).
027UFFELIE, O. F., Pharm. Weekbl. 87, 646 (1952).
028WALASZEK, E. J., PIRCIO, A., J. Am. Pharm. Ass. 41, 270 (1952).
029SANTAVY, F., CERNOCH, M., Chem. Listy 46, 81 (1952).
030VOLKE, J., FORTOVA, V., Dissertation, Charles University, Praha, 1951.
031MATSUMUTO, K., J. pharm. Soc. Japan 72, 1393, 1396, 1398 (1952).
032JINDRA, A., JUNGR, V., ZYKA, J., Cs. farm 1, 177 (1952).
033RUSCONI, Y., MONNIER, D., WENGER, P. E., Anal. Chim. Acta 5, 222 (1951).
034DEROBERT, L., TRUFFERT, L., LEBRETON, R ., Ann. Med. leg. Criminal 31, 47 (1951); C.A. 45, 7180e (1951).
035WENGER, P. E., MONNIER, D., SCHMIDGALL, H., Helv. Chim. Acta. 35, 1108, 1192, 2242 (1952).
036VALENTIN, H., FRANCK, R., Pharm. Ztg. 81, 943 (1936). FRANCK, R., Arch. Pharm. 275, 125 (1937).
037MERZ, K. W., FRANCK, R., Arch. Pharm. 275, 345 (1937).
038KONDO, H., J. Pharm. Soc. Japan 57, 218 (1937).
039LEVI, J, R., CASTELLI, F., Anales farm. bioquim. 11, 6 (1940).
040BJÖRLING, C. O., Svensk Farm. Tidskr. 48, 137, 161 (1944).
041LJUNBERG, S., S vensk Farm. Tidskr. 50, 197, 237 (1946).
042MUHLEMANN, H., TOBLER, R., Pharm. Acta Helv. 21, 34, 65 (1946).
043LA ROCCA J., BURLAGE, H. M., J. Elisha Mitchell Sci. Soc. 61, 220 (1945).
044REIMER, F., GOTTLIEB, K. R., CHRISTENSEN, V. Aa., Quart. J. Pharm. Pharmacol. 20, 99 (1947).
045BJORLING, C. O., Acta Chem. Scand. 1, 392 (1947).
046BROWN, N. G., KIRCH, E. R., WEBSTER, G. L., J. Am. Pharm. Ass. 37, 24 (1948).
047LASSLO, A., WEBSTER, G. L., J. Am. Pharm. Ass. 39, 193 (1950).
048KLEE, F. C., KIRCH, E. R., J. Am. Pharm. Ass. 42, 146 (1953).
049STOLMAN, A., STEWART, C. P., Analyst 74, 536 (1949).
050GUANI, B. P., GANGULY, P. B., J. Ind. Chem. Soc. 19, 453 (1942).
051EVANS, W. C., PARTRIDGE, M. W., Quart. J. Pharm. Pharmacol. 21, 126 (1948).
052MUNIER, R., MACHEBCEUP, M., Bull. soc. chim. biol. 31, 1144 (1949).
32, 192 (1950).
053BRINDLE, H., CARLESS, J. E., WOODHEAD, H. B., J. Pharm. Pharmacol. 3, 793 (1951).
054ROMEIKE, A., Die Pharmazie 7, 496 (1952).
055ERBEN, J., Dissertation, Charles University, Praha, 1949.
056UNGERER, E. V., Kolloid Z. 36, 228 (1925).
057OBERST, M. S., Lab. Clin. Med. 24, 318 (1938).
058FISCHER, R., CHALUPA, L., , Mikrochemie 34, 257 (1949); 35, 63 (1950).
059MCINTOSH, B. J., KELSEY, F. E., GEILING, E. M. K., J. Am. Pharm. Ass. 39, 512/195.
060BJ?RLING, C. O., BERGGREN, A., J. Pharm. Pharmacol. 5, 169 (1953). BERGGREN, A., BJORLING, C. O., J. Pharm. Pharmacol. 5, 615 (1953).
061ADAMS, B. A., HOLMES, E. L., J. Soc. Chem. Ind. 54, 17 (1935).
062NACHOD, F. C., Ion Exchange 353, New York 1949.
063APPLEZWEIG, N., J. Am. Chem. Soc. 66, 1990 (1944).
064APPLEZWEIG, N., RONZONE, S. R., Ind. Eng. Chem. 38, 576 (1946).
065APPLEZWEIG, N., ref. NACHOD, F. C., Ion Exchange 354, New York 1949.
SULLIVAN, M. J., MARTIN, G. J., Amer. J. Pharm. 122, 48 (1950).
066KINGSBURY, A. W., MINDLER, A. B., GILWOOD, M. E., Chem. Eng. Process 44, 497 (1948).
067WINTER, J. C., KUNIN, R., Ind. Eng. Chem. 41, 460 (1949).
068SAUNDERS, L., SRIVASTAVA, R., J. Chem. Soc. 1950, 2915.
069SAUNDERS, L., SRIVASTAVA, R., J. Chem. Soc. 1952, 2111.
070HUYCK, L., Amer. J. Pharm. 122, 228 (1950).
071SAUNDERS, L., J. Pharm. Pharmacol. 5, 569 (1953).
072BUCHI, J., FURRER, F., Arzneim. Forsch. 3, 1 (1953).
073GRANT, E. W., HILTY, W. W., J. Pharm. Am. Ass. 42, 150 (1953).
074SUSSMAN, S., MINDLER, A. B., WOOD, W., Chem. Ind. 57, 455, 549 (1945).
075JAMPOLSKAJA, M. M., Apt. delo 2, 17 (1953).
076JINDRA, A., POHORSKY, J., J. Pharm. Pharmacol. 2, 361 (1950); 3, 344 (1951); Cas. ces. l?k?rn. 63, 57 (1950).
077JINDRA, A., RENTZ, J., Cs. farm. 1, 625 (1952).
078SAUNDERS, L., J. Pharm. Pharmacol. 5, 569 (1953).
079JINDRA, A., RENTZ, J., J. Pharm. Pharmacol. 4, 632 (1952).
080JINDRA, A., RENTZ, J., Cs. farm. 1, 625 (1952).
081JINDRA, A., MOTL, O., Cs. farm. 1, 632 (1952).
082JINDRA, A., MOTL, O., Cs. farm. 2, 190 (1953).
083MOTL, O., Cs. farm. 1, 630 (1952).
084BAGGESGAARD-RASMUSSEN, H., FUCHS, D., LUNDBERG, L., J. Pharm. Pharmacol. 4, 566 (1952).
085LEVI, L., FARMILO, Ch. G., Can. J. Chem. 30, 793 (1952).
086GUNDERSEN, F. O., HEIZ, R., KLEVSTRAND, R., J. Pharm. Pharmacol. 5, 608 (1953).
087KRESSMANN, I. R. E., Mfg. Chemist 23, 93, 149, 194, 241 (1952).
088JINDRA, A., MOTL, O., Die Pharmazie 8, 547 (1953).

