ABSTRACT
HISTORICAL REVIEW
EXPERIMENTAL RESULTS AND DISCUSSION
Physicochemical Characterization of the Derivative
ACKNOWLEDGEMENT
Microchemical Identification of Dromoran
Author: Leo Levi
Pages: 43 to 59
Creation Date: 1955/01/01
When 3-hydroxy-N-methylmorphinan interacts with chloroplatinic acid in aqueous ethanol a complex having the composition [C 17H 23NO.H] 2.[PtCl 6] precipitates on standing. Product yields are dependent upon the pH of the medium and availability of the alkaloidal cation is most likely the rate-controlling step in the metathetical process of complex formation. The reaction is highly sensitive and under suitable conditions as little as 1γ of the amine can still be readily detected. It fails with opium alkaloids of closely related structures which emphasizes its value as a specific microchemical method for identifying the synthetic narcotic. The X-ray diffraction pattern, ultraviolet and infrared absorption spectra, the dissociation constant and solubility behaviour of the complex are reported and these data utilized to interpret characteristic structural features of the alkaloid and its derivative. The constants thus derived are also of value for the determination of minute amounts of the narcotic drug.
TABLE I
Synthesis of N-Methylmorphinan according to Grewe
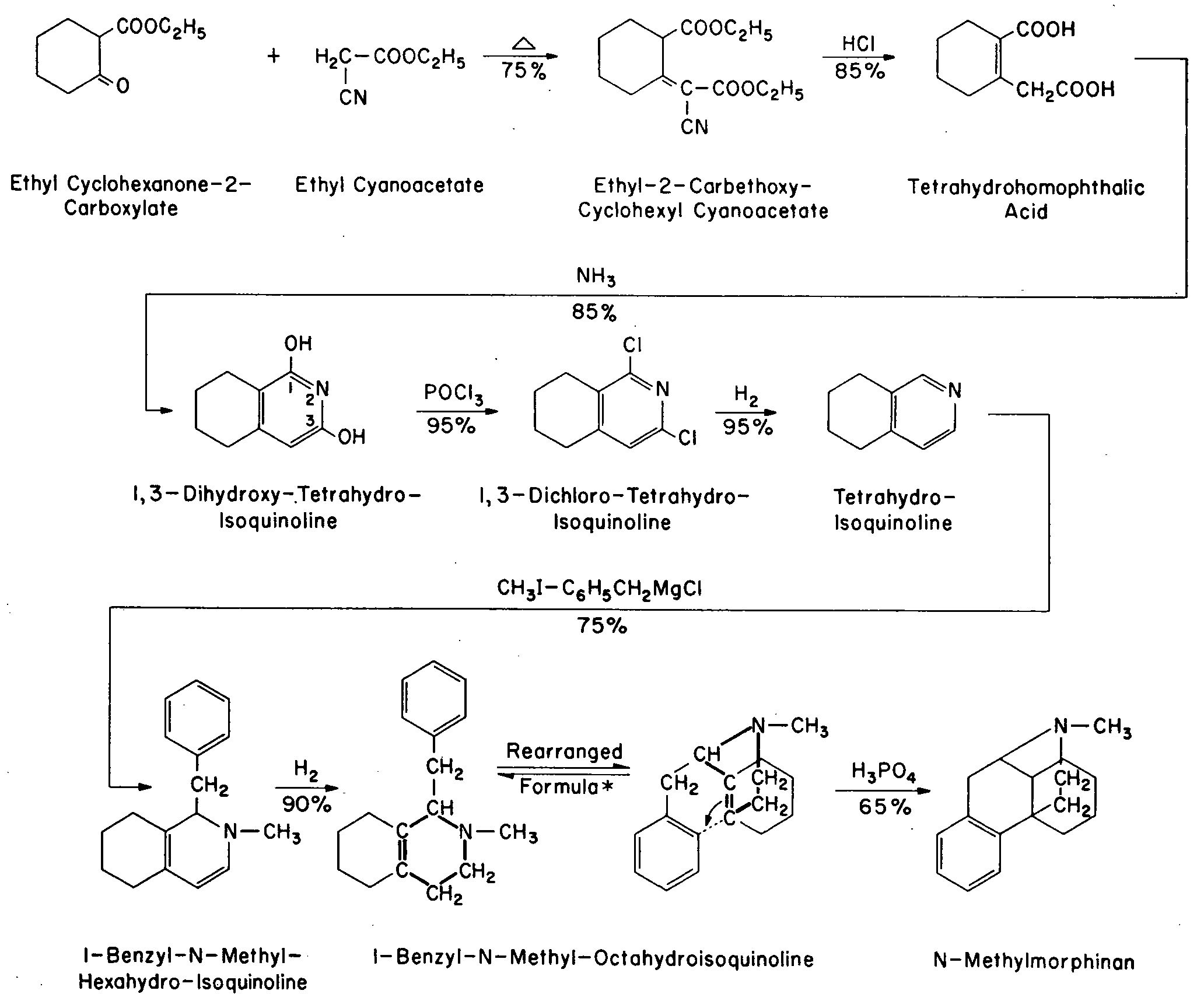
* Heavy lines indicate heterocyclic unit of the molecule and arrow shows mode of ring closure involving the benzene nucleus and the unsaturated fused ring system to form an angular phenyl group. As far as the author is aware, no analogy can be found in the literature for this interesting reaction, which comes close to Robinson's theoretical concepts regarding the synthesis of morphine by the opium poppy [4la] .
On 21 October 1946, an important paper was presented by R. Grewe to a group of distinguished chemists assembled in the city of Göttingen. After seven years of strenuous research on the synthesis of morphine [39] , [40] the famous scientist had succeeded in preparing a compound whose structure came closer to the model than any other synthetic known at that time. The compound was "2-methyl-5,9-tetramethylene-6,7-benzo-2-aza-bicyclo-(3,3,1)-nonene-6".Grewe called it briefly "Morphan", so as to express more strikingly its fundamental resemblance to morphine, and regarding the importance of the chemical process itself he made, the following prediction : " Man darf vermuten, dass die neue synthetische Methode die Darstellung zahlreicher Substanzen vom Morphin-Typ erlauben wird, die sich durch eine besondere pharmakologische Wirkung auszeichnen und dass damit ein neuer Weg zu synthetischen Arzneimitteln eröffnet wird."
A schematic representation of the ingenious synthesis of the molecule together with yields realized in the various steps is given in Table I.
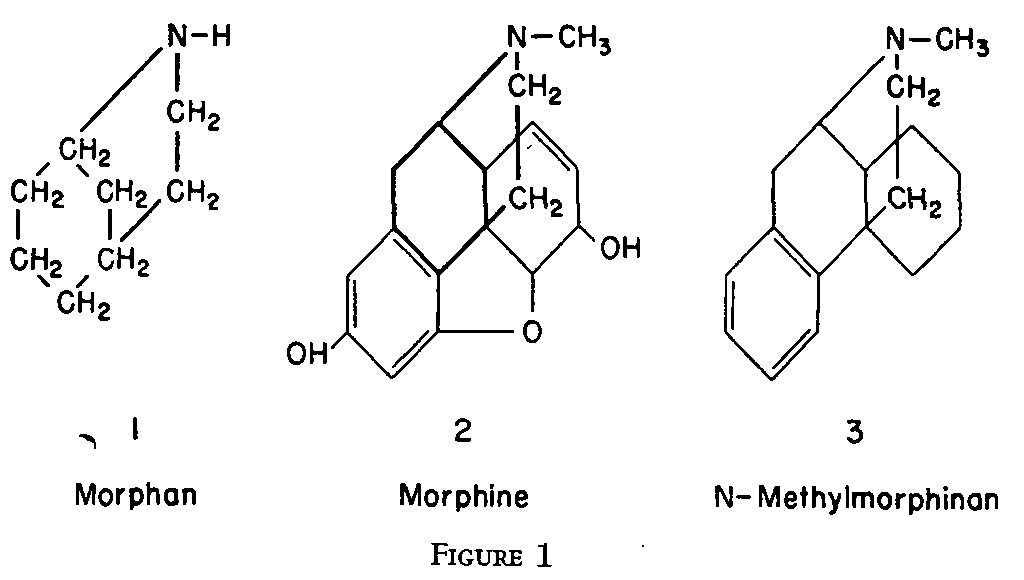
Just about the time Grewe reported the results of his brilliant investigations, J. A. Barltrop in England was making a significant contribution to the classical problem. Believing that formation of the nitrogen-containing meta-bridged ring system present in morphine - see Fig. 1, heavy lines - was likely to prove one of the most difficult stages of the process he set out to synthesize this particular portion of the molecule and by cyclizing β - diethylaminoethyl derivatives of ethyl-cyclohexanone carboxylate with 1-alkyl-β-tetralones he was able to isolate a series of compounds - bicyclo-(3,3,1)-2-azano-nanes - showing the desired ring system [1] . * Like Grewe, Barltrop was anxious to " save difficulties with the cumbrous and obscure bicycloazanomenclature " and following a suggestion of Sir Robert Robinson, Head of Oxford University's Dyson Perrins Laboratory, he gave the 9-membered N-bridged ring system the trivial name " Morphan ".
When Grewe's paper appeared in Die Naturwissenschaften [38] Sir Robert immediately communicated with the German scientist, proposing that the name " morphan " be retained for Barltrop's cyclic amines and his (Grewe's) ring system be designated as "morphinan". This suggestion was endorsed by Grewe [40] .
The structural resemblance of the various molecules referred to is graphically illustrated in Fig. 1. Inspection of these formulae shows that four operations would still be required for the conversion of N-methylmorphinan to morphine - namely, the introduction of a phenolic hydroxyl group, a secondary alcoholic hydroxyl group, an olefinic double bond and an oxygen bridge linking the aromatic with the hydro genated ring. So far, only one of these operations has been successfully carried out - namely, the introduction of a phenolic hydroxyl group into the 3-position. Schnider and Grussner, working at the research laboratories of Hoffmann LaRoche, Inc., Basle, Switzerland, synthesized 3-hydroxy-N-methylmorphinan [65] * as shown in Table II and later Schnider and Hellerbach [66a] prepared the compound as illustrated in Table II a.
Barltrop did not isolate the parent compound of this series, but only derivatives thereof. The unsubstituted, bicyclic secondary amine was synthesized two years later, independently and almost simultaneously, via lactamization of cis-3-aminocyclohexaneacetic acid and subsequent catalytic reduction of the resulting lactam, by Cronyn at the University of California, Berkeley, Cal. [14] and by Ginsburg at Rehovoth's Weizmann Institute of Science in Israel [31] .
The structural similarity of the hydroxylated derivative to morphine was soon found to be paralleled by striking similarities in pharmacological behaviour, the analgesic action of the compound proving equal and even superior to that of the natural opium alkaloid [3, 6, 10, 11, 15, 28, 29, 32, 39a, 41, 42, 43, 45, 47, 48, 50, 59, 63, 64, 67, 68, 70, 74, 78, 79, 81, 83] . However, the extensive clinical evaluations also revealed that, like morphine, the compound - tentatively designated by Hoffmann LaRoche as NU-2206 (NU standing for Nutley, location of their U.S.A. headquarters, and 2206 being the laboratory number of the product) [66 b] - was to be considered an addiction-producing drug. Single doses administered subcutaneously or intravenously produced euphoria and afforded relief of the withdrawal syndrome when given at the height of abstinence from morphine. Also, following abrupt withdrawal after prolonged administration definite signs of abstinence became apparent [28, 32, 45, 48, 63, 78, 79] .
In the light of these findings the United States Government, pursuant to article I, paragraph 1, of the Paris Protocol, addressed on 17 October 1950 to the Secretary-General of the United Nations a notification, along with pertinent clinical reports from its Public Health Hospital at Lexington, Ky., stating that the drug which may be used for medical and scientific purposes was to be considered liable to the same kind of harmful effects as the drugs specified in article I, paragraph 2, of this Convention. The Secretary-General of the United Nations duly transmitted this communication to the other States Parties of the Protocol, the Commission on Narcotic Drugs and the World Health Organization, whose Expert Committee on Drugs Liable to Produce Addiction considered all relevant data at its third session held in Geneva, 7-12 January 1952. The Committee expressed the opinion " that 3-hydroxy-N-methylmorphinan, because it [1] produces morphine-like euphoria, [2] will suppress the abstinence phenomena of a known morphine addiction and [3] will sustain a morphine addiction, must be considered an addiction-producing drug comparable to morphine" [79] and that, therefore, the regime laid down in the 1931 Convention for the drugs specified in article I, paragraph 2, group 1, should be applied to the synthetic alkaloid and its salts. The recommendation was referred back to the Secretary-General of the United Nations, who notified the States Parties accordingly. Thus, the drug 3-hydroxy-N-methylmorphinan came to be placed under international control.* It was marketed by Hoffmann LaRoche as the hydrobromide salt under the trade name of Dromoran®.
* Two other methods were reported by these workers at the same time
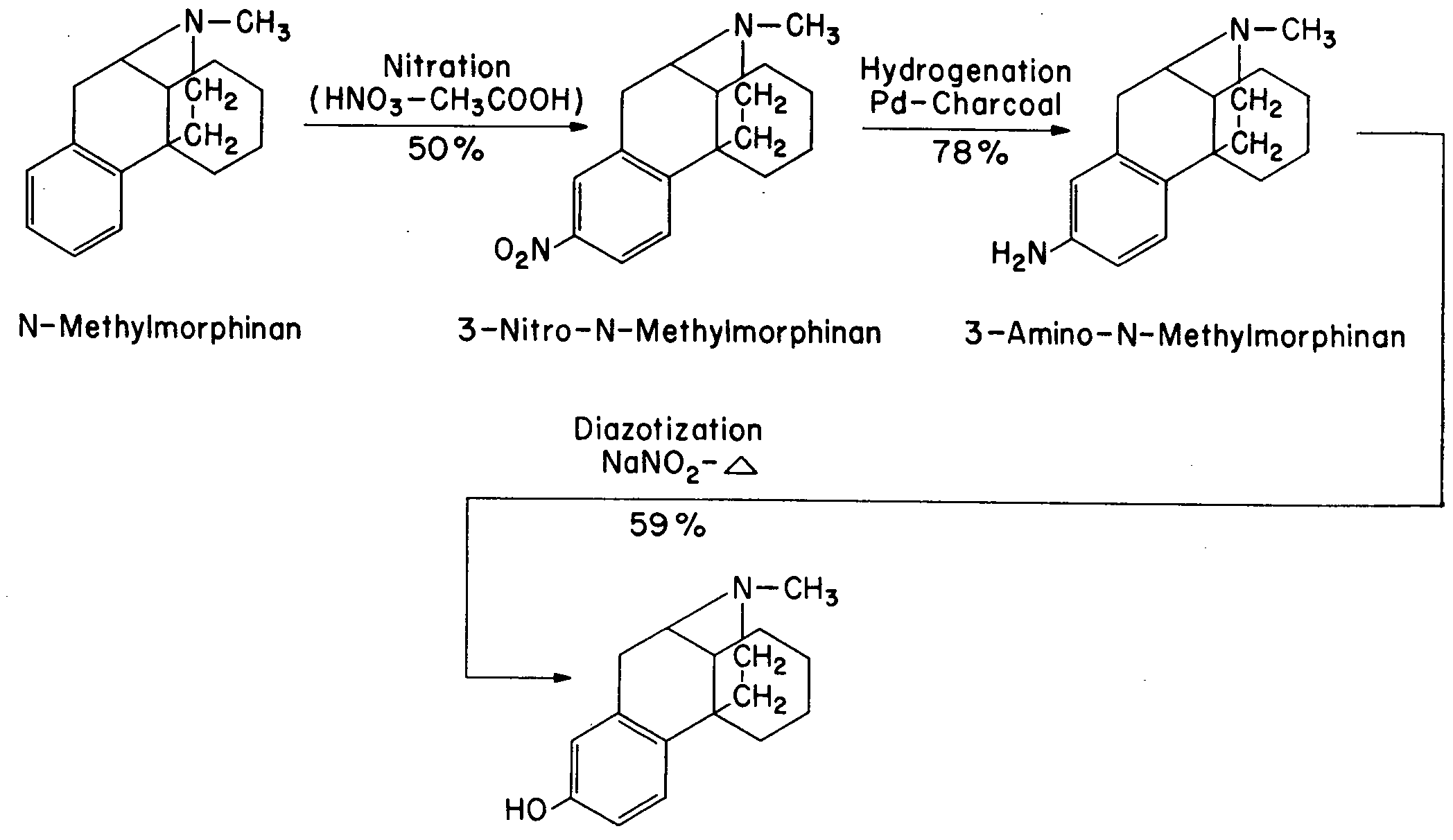
It should be emphasized at this point that chemically Dromoran was a racemoid ( d,l-compound) and since such a substance is biologically always less active than either its l- or d-configuration (natural morphine is a levo isomer) further researches were carried out at the Hoffmann LaRoche laboratories to resolve the product into its optical antipodes. Schnider and Grussner realized this process by treating the base with D-tartaric acid and separating the optically active salts on the basis of their different solubilities in water [66] . Photomicrographs of the two isomers as published in Helvetica Chimica Acta are reproduced in Fig. 2.
* Provisionally, the compound had already been put under the regime laid down in the 1931 Convention for the drugs in group 1 following a resolution-taken in pursuance of article 2 of the Protocol by the Commission on Narcotic Drugs, during its sixth session held in New York, 10 April-24 May 1951 [20] .
Clinical evaluations soon showed that the dextro isomer exhibited no pain-relieving properties while the levo compound displayed considerably greater analgesic action than did the racemate [29] , [45] . Following these observations Hoffmann LaRoche discontinued manufacturing d,l-Dromoran Hydrobromide and began to market l-Dromoran Tartrate (Levo-Dromoran, Brand of Levorphan Tartrate) instead.
In Canada, Dromoran and Dromoran compounds were already included in the Schedule of the Opium and Narcotic Drug Act in 1950 (Order in Council PC 1578, dated 28 March 1950) by the Division of Narcotic Control acting in accordance with the international treaty known as the 1948 Paris Protocol.* Sensitive microchemical methods of identification and characterization thus became imperative and research along these lines was immediately begun in the federal laboratory.
It soon became apparent that, unlike morphine, Dromoran was extremely reluctant to yield crystalline derivatives which would prove of value to the forensic chemist. Of 224 reagents tested, 222 gave amorphous precipitates and only two produced characteristic microcrystalline complexes suitable for the identification of minute amounts of the narcotic. These two reagents were Reinecke's Salt and chloroplatinic acid. The characterization of Dromoran (and other narcotics) with Reinecke's Salt has already been reported [54] and it is the purpose of this paper to present a study of the reaction of the clinically important drug with chloroplatinic acid.
* The nomenclature of these compounds may easily lead to confusion and it is here reproduced for reference purposes from a paper by Braenden and Wolff [5] .
Levorphan*: (-)-3-hydroxy-N-methylmorphinan, also described as l-3-hydroxy-N-methylmorphinan
Synonyms : Dromoran,
Laevo-Dromoran Tartrate
Racemorphan *: (±)-3-hydroxy-N-methylmorphinan, also described as rac. 3-hydroxy-N-methylmorphinan
Synonym : Cetarin (The name Dromoran was previously given to Racemorphan, but is now used for Levorphan).
Levomethorphan *: (-)-3-methoxy-N-methylmorphinan, also described as l-3-methoxy-N-methylmorphinan
Racemethorphan *: (±)-3-methoxy-N-methylmorphinan, also described as rac. 3-methoxy-N-methylmorphinan
* Proposed International Non-proprietary Name

Preparation of the Derivative
To a solution of 0.005 moles (1.7365 g) of d, l-3-hydroxy-N-methylmorphinan hydrobromide in 150 ml of distilled water was added, slowly and with stirring, a solution of chloroplatinic acid, prepared by dissolving 0.005 moles of the salt (2.59 g of H 2PtCl 6.H 20) in a mixture of 75 ml of 1 N hydrochloric acid and 75 ml of ethanol. The bulky, microcrystalline precipitate which formed gradually was filtered off after 12 hours, standing and dried in vacuo over phosphorous pentoxide. (Weight of product = 2.271 g or 98.24% theoretical yield.)
The yellowish-coloured compound - practically insoluble in water and organic solvents - was purified by reprecipi- tation with ethanol from 50% aqueous acetone solution. Two such recrystallizations yielded a product, which after drying over P 2O 5 proved sufficiently pure for physicochemical characterization.
Qualitative and Quantitative Analyses
The complex thus prepared could be dried at 100°C. for two hours without suffering loss of weight or colour. Karl Fisher titrations showed that it did not contain any water of crystallization. It decomposed to a dark brownish liquid from 235-238°C. as determined with a Fisher-Johns melting-point apparatus adjusted to a heating rate of 5°C. per minute. When reacted with potassium iodide a brownish-red colouration was observed (platinum) [76] , and on treatment with silver nitrate a flocculent precipitate formed immediately (halide ions). No colour formation was noted, however, when a sample was reacted with chlorine water in dilute sulphuric acid (absence of bromine) [76] . Also, on addition of aqueous potassium chloride to a solution of the complex in dilute acetone a yellow precipitate formed almost immediately (presence of PtCl 6= ions)* [76] , and on reaction with Froehde's reagent a dark blue-green colouration developed immediately (3-hydroxy-N-methylmorphinan) [21] .
PtCl 4 gives no precipitate with potassium salts - or at least only after long standing[76] .
Quantitative analyses indicated that the composition of the complex was in agreement with the formula [C 17H 23NO.H] 2. [PtC l6]. The alkaloid and metal were determined following decomposition of the complex with hydrogen sulfide and the halide was estimated as silver chloride.
Determination of the Metal
About 0.1 g of the derivative was weighed out accurately and dissolved with gentle heat in 100 ml of 2% aqueous hydrochloric acid. A slow stream of hydrogen sulfide was passed through the hot solution and the precipitated platinum sulfide-filtered off after 12 hours' standing - ignited to constant weight in a muffle furnace.
Determination of the Narcotic
The filtrate collected from the previous assay was evaporated to a volume of about 20 ml, made alkaline with dilute sodium carbonate solution and extracted with successive 20-ml portions of chloroform. Removal of the alkaloid from the aqueous system was considered complete when the residue obtained after evaporation of five drops of the organic layer did no longer respond to Fulton's phosphomolybdic acid test [30] . After washing twice with 3-ml portions of distilled water the extract was placed on a steam bath and following removal of the solvent dried at 100 °C. The residue was weighed as 3-hydroxy-N-methylmorphinan (M.P. 248-251 °C.).
Determination of the Halide
Like morphine, Dromoran and Dromoran compounds were found to reduce silver salts to metallic silver and hence when reacting the complex with silver nitrate the precipitated silver chloride was always contaminated with colloidal silver. It was found possible to remove the metal from the system via conversion to silver sulphide and subsequent decomposition of the silver salt by means of nitric acid.
About 100 mg of the finely powdered complex were weighed out accurately into a 100-ml beaker. An equal quantity of sodium sulphide was added and the mixture triturated intimately with 5 ml of distilled water. During this operation black platinum sulphide precipitated while the liberated halide was converted to sodium chloride. After diluting to a volume of about 50 ml, the preparation was placed on a steam bath for half an hour to insure completion of the reaction and, following replacement of the water lost by evaporation, acidified with 5 ml of 10% sulphuric acid in order to convert all excess sodium sulphide to hydrogen sulphide. The precipitated platinum sulphide was filtered off-along with some colloidal sulphur which formed on standing-and to the filtrate was added a solution of 0.3 g of silver nitrate in 25 ml of water containing 1 ml of nitric acid. After 12 hours the supernatant liquid was decanted through a Whatman No. 42 filter paper and, using about 10 ml of water, any precipitate carried over was carefully transferred back to the bulk of the residue. Three ml of concentrated nitric acid were added to it and the suspension was digested on a steam bath for about 15 minutes in order to convert all silver sulphide contaminating the silver chloride to soluble silver nitrate.
|
Platinum |
Dromoran |
Chlorine | |||
|---|---|---|---|---|---|
|
Found |
Theory |
Found |
Theory |
Found |
Theory |
| 21.03 | 21.10 | 55.09 | 55.67 | 22.79 | 23.00 |
| 20.84 | - | 55.38 | - | 22.36 | - |
After diluting with 75 ml of distilled water the mixture was placed in the dark overnight, the silver chloride filtered off in subdued light, washed with a little ethanol and carbon disulphide to remove free sulphur (see equation), dried at 110°C. and weighed. The experimental data are recorded in Table III.
Ultraviolet Absorption
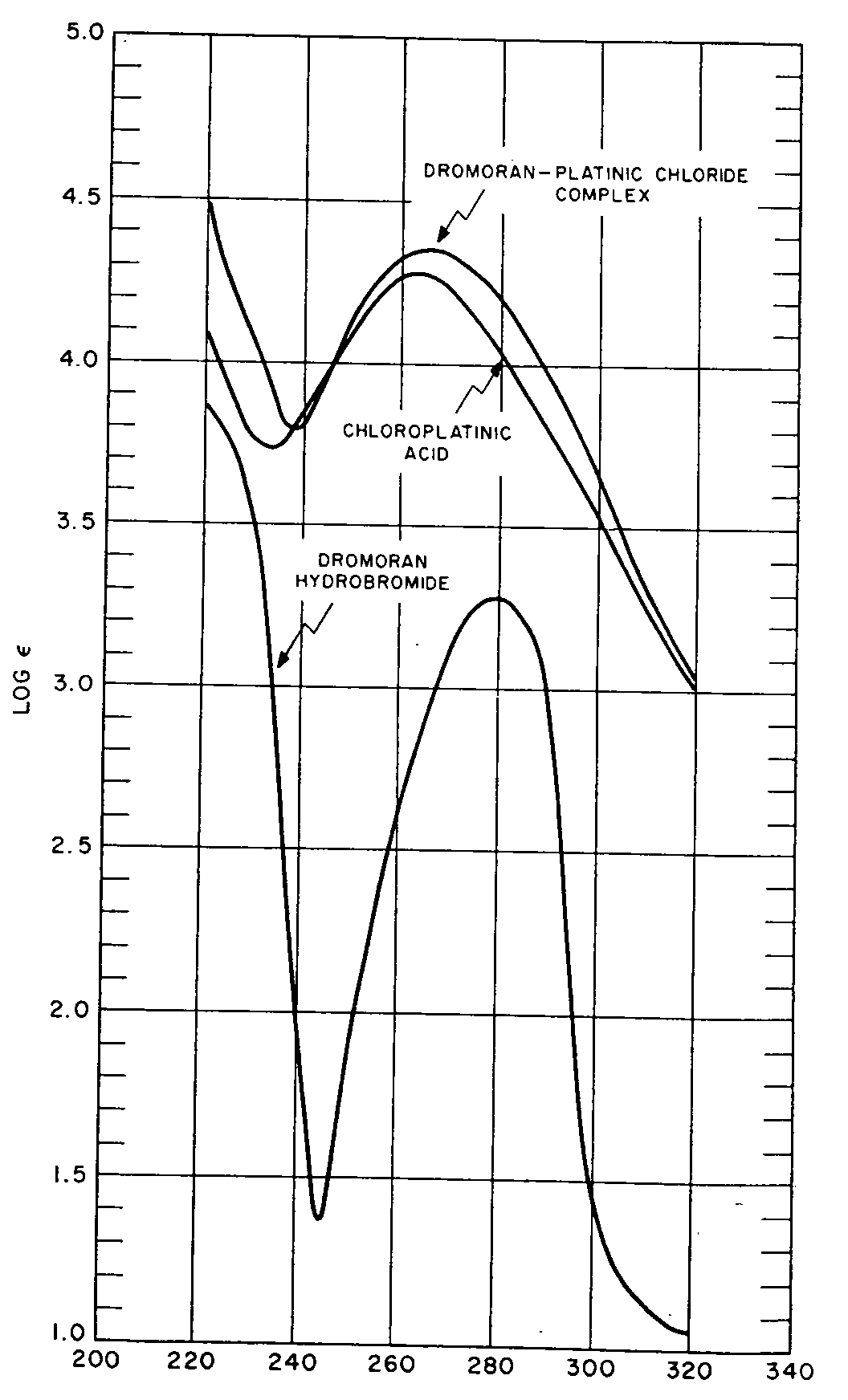
A 0.00002974-molar aqueous solution of the complex was prepared (0.0275 g dissolved with gentle heat in 1000 ml of distilled water) and its ultraviolet absorption measured in a Beckman Model DU Spectrophotometer equipped with photomultiplier tube. Similar determinations were made on the alkaloidal salt and the inorganic reagent. The experimental data are recorded in Fig. 3 and Table IV.
|
Compound |
Concentration (g/l) |
max (mμ) |
max. |
min (mμ) |
min. |
|
Dromoran hydrobromide . |
0.1051 | 279 | 1,881 | 245 | 23.4 |
|
Dromoran platinic chloride |
0.0275 | 265 | 22,126 | 238 | 6,321 |
|
Chloroplatinic acid. . . . . . .. |
0.0172 | 262 | 19,117 | 232 | 5,649 |
Because of the deliquescent nature of chloroplatinic acid, its ultraviolet absorption could not be measured directly on accurately weighed samples of the compound. Good results were obtained, however, by analysing, prior to UV-determinations, 50-ml aliquots of an approximately 0.0025-molar aqueous solution of the salt for platinum (precipitation by means of hydrogen sulphide) and chlorine (precipitation by means of silver nitrate). Duplicate determinations showed the presence of 20.55 mg of platinum or the equivalent of 43.19 mg of anhydrous chloroplatinic acid and 22.21 mg of chlorine or the equivalent of 42.80 mg of anhydrous chloroplatinic acid per sample. The solution was therefore considered to contain 0.8599 g of anhydrous H 2PtCl 6 per litre. Ten ml of this preparation were diluted to 500 ml with distilled water to make a 0.00005-molar solution whose ultraviolet absorption was measured.
Comparison of the spectral curves reproduced in Fig. 3 shows that the complex exhibits an absorption pattern considerably different from that of the alkaloidal salt used for its synthesis, which observation indicates that formation of the derivative may be used advantageously for the characterization of the narcotic. One can also see that the inorganic salt, by virtue of the fact that it absorbs strongly in the ultraviolet region and comprises 44.34% by weight of the compound, affords a decisive contribution to the ultraviolet absorption characteristics of the complex. It should further be noted-see Table IV-that the molar extinction coefficient of the complex exceeds the sum values of the molar absorptions exhibited by the alkaloidal salt and the reagent. Evidently, the process of complex formation is associated with considerable electronic exaltation of the molecular system.
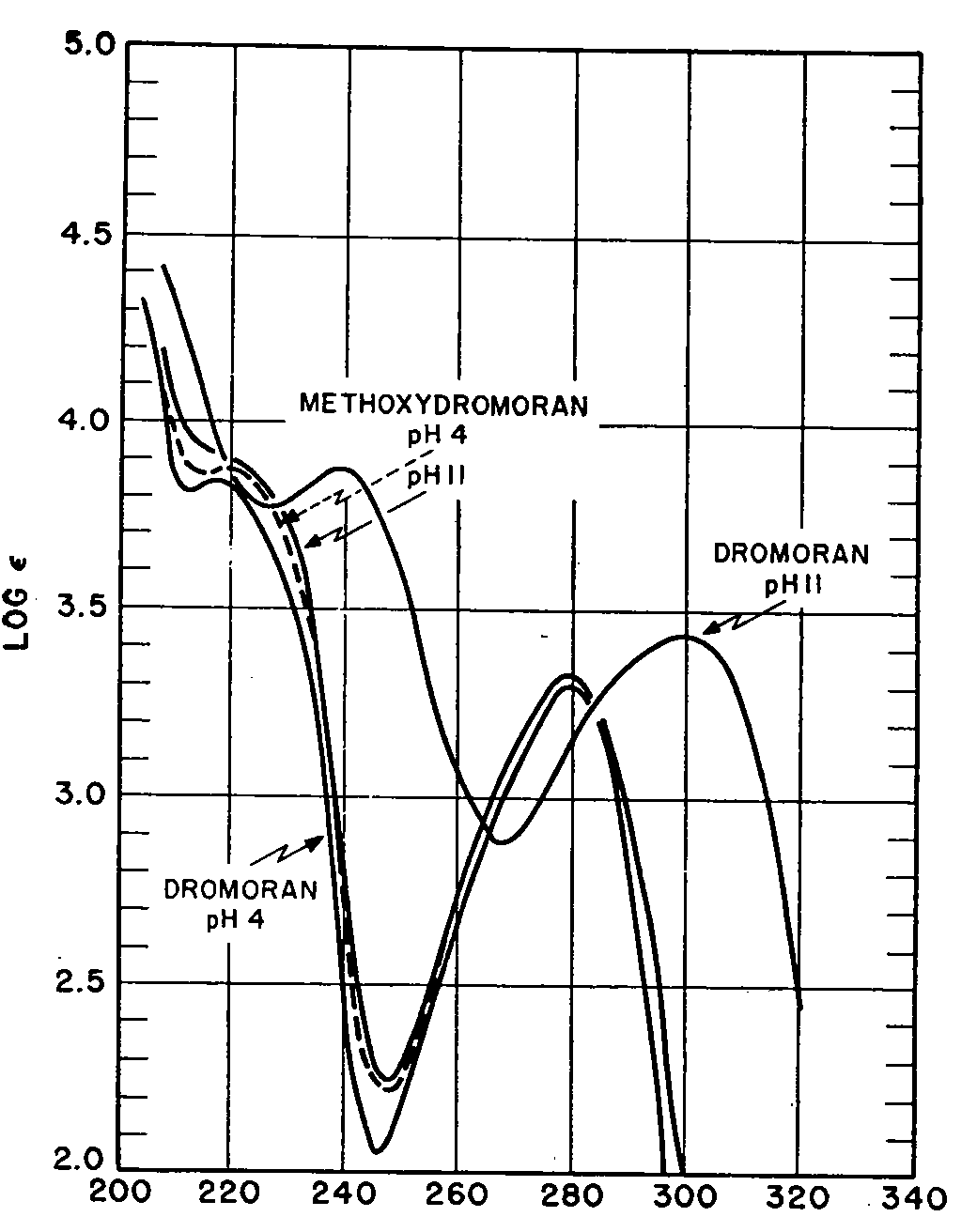
Further ultraviolet data were assembled by isolating the narcotic from the complex in accordance with the procedure described and measuring its absorption at two different pH values. The experimental results are recorded in Figs 4 and 5 and characteristic constants of the compound listed in Table V along with similar data on the methylated analogue, 3-methoxy-N-methylmorphinan, and the closely related opium alkaloids morphine and codeine.
The narcotics dissolved readily in buffer systems at pH 4 and both morphine and codeine solubilized without difficulty at pH 11. Dromoran and Methoxydromoran, however, were found to be practically insoluble in aqueous alkaline media and their UV-absorptions were measured by first dissolving representative samples in 5 ml of ethanol. The buffer systems employed were those described by Morgan [57] and they also served as blanks in all of the measurements reported.

It can be seen that at a pH of 4 Dromoran exhibits two maxima (218 and 279 mμ) both of which are displaced by 21 mμ toward higher wavelengths at pH 11 and similar bathochromic shifts move its minima, occurring at 212 and 246 mμ, to 226 and 267 mμ, respectively. Morphine also displays such pH-dependence of its ultraviolet absorption, the maxima occurring at 210 and 286 mμ as well as the minimum observed at 262 mμ shifting progressively toward higher wavelengths (by 8, 12 and 17 mμ respectively) as the pH of the system is increased from 4 to 11. Codeine (methylated morphine) does not show this phenomenon and, likewise, Methoxydromoran (3-methoxy-N-methylmorphinan) displays neither hypso-nor bathochromic absorption shifts with change in pH. Evidently, the phenolic hydroxyl group present in Dromoran and morphine (production of phenolate ions) is responsible for this characteristic ultraviolet behaviour pattern. It should also be noted that one of the maxima (216 mμ) and one of the minima (220 mμ) exhibited by Methoxydromoran at pH 4 are no longer detectable at pH 11. These bands are to be considered characteristic of the ionic species of the alkaloid. They disappear at higher pH values when dissociation of the base is suppressed and only molecular absorption takes place. No such phenomenon is shown by codeine because this compound may ionize in both acidic and basic media (presence of alcoholic hydroxyl group).
Further interesting observations can be made by comparing the molar extinction coefficients of the four narcotics at the two pH values recorded. Thus, when changing the hydrogen ion concentration of the medium from 4 to 11, one of the maximal molar extinction coefficients of morphine (286 mμ) is increased by 1,036, the other (210 mμ) reduced by 2,747 and the minimal molar extinction (262 mμ) increased by 1,099 units. In Dromoran these hyperchromic shifts are also evident but of considerably smaller magnitude (about 600-700 units) because the molecule lacks the 7,8-ethylenic linkage and hyperconjugation via an alcoholic hydroxyl group cannot take place. It is also to be noted that codeine, unlike morphine, shows a general depression of its absorption at pH 11 (Δ⊂ = - 6 to -454); while Methoxydromoran displays, under comparable experimental conditions, but a slight exaltation of its molar extinction (8 to 28). Thus, the marked auxochromic effects of methoxyl substituents, phenolic and alcoholic hydroxyl groups on the ultraviolet absorption of these compounds become very apparent.
From the data recorded in Table V it also follows that the spectral behaviour of the narcotics analysed is strongly influenced by the nature of the solvent. Thus, in ethanol, Dromoran exhibits one of its maxima and both of its minima, observed at 279, 212 and 246 mμ respectively in aqueous media of pH 4, at longer wavelengths (283, 218 and 251 mμ respectively) and shows generally more marked absorptions (Δ ⊂ =100-2,658). Methoxydromoran displays in ethanol a maximum and a minimum (289 and 287 mμ respectively) not present in aqueous systems at pH 4 or 11, and with the exception of its minimal absorption at 248 mμ, which appears to be practically independent of pH and solvent, its molar extinction is generally also higher in ethanol (Δ⊂= 244 - 521) than in aqueous media. These UV characteristics are less pronounced in the spectra of morphine and codeine, which compounds exhibit both their maxima and minima at practically identical positions and show comparable molar extinctions in both ethanol and slightly acidic media. The only conspicuous feature to be noted is the absence of a maximum in the ethanol spectrum of morphine at 210 mμ where marked absorption occurs when the compound is examined in aqueous systems at pH 4 and 11.
The phenomena observed are not readily explained and a considerable amount of fundamental investigations into the effects of hydrogen bonding, temperature, conjugation and dipole moment on the spectra of these compounds and their analogues would have to be done before providing even tentative interpretations. Unfortunately no systematic collection of data of this kind nor of the type here reported is at present available for narcotics and related alkaloids. It would, no doubt, prove of great practical value to the forensic chemist because such data form the basis of sensitive differential methods of analysis, yield a considerable amount of information regarding the chemical constitution of narcotic compounds and are of cardinal importance for elucidating the mechanisms of their reactions [4, 7, 8, 9, 12, 13, 16, 17, 18, 19, 22, 23, 23a, 24, 25, 26, 27, 33, 34, 36, 37, 44, 46, 49, 51, 52, 53, 55, 56, 57, 58, 60, 61, 62, 69, 71, 72, 73, 75, 77, 82] . It is contemplated to carry out such research at the federal laboratory and publish a survey of this nature as continuation of the work of Oestreicher, Farmilo and Levi comprising the ultraviolet spectra of narcotics and related compounds [61] .
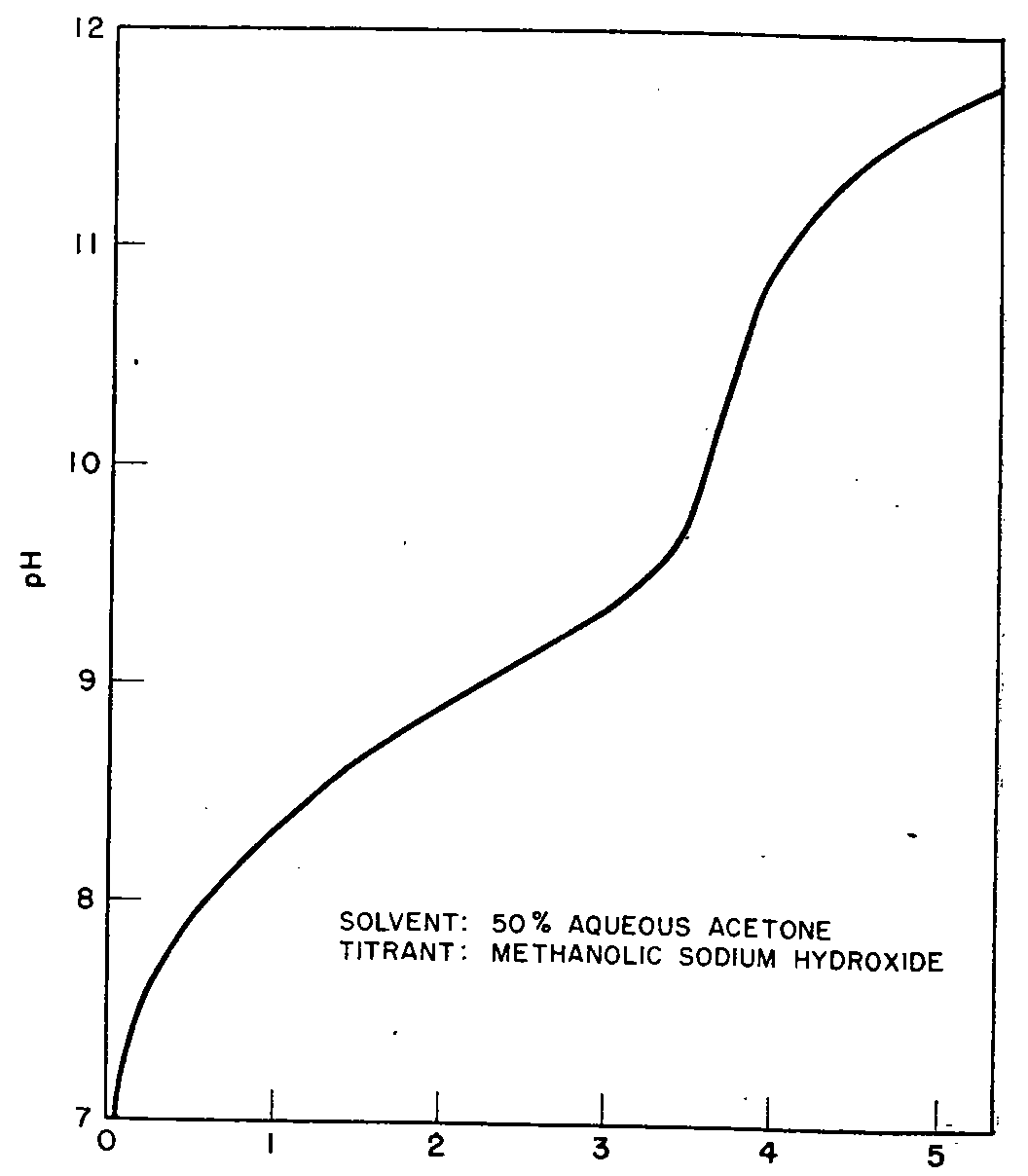

Potentiometric Titration
Because of its insolubility in water the complex could not be analysed by aquametric titration. Quantitative estimation was possible, however, by means of 0.05 N alcoholic sodium hydroxide, in a solvent system consisting of equal parts of water and acetone. Two moles of titrant were found to be consumed per mole of complex in accordance with the equation:
[Dromoran. H] 2[PtCl 6] + 2 NaOH →
2 Dromoran H+ + 2 OH - + Na 2PtCl 6.
The titration curve showed a sharp break at the equivalence point-see Fig 6-and indicated a recovery of 99.87%. Also from the experimental data a dissociation constant (pK B) of 5.68 was calculated for the narcotic. This value agrees closely with that derived by Levi and Farmilo by analysis of the Dromoran-Reineckate complex [54] .
Infrared Absorption
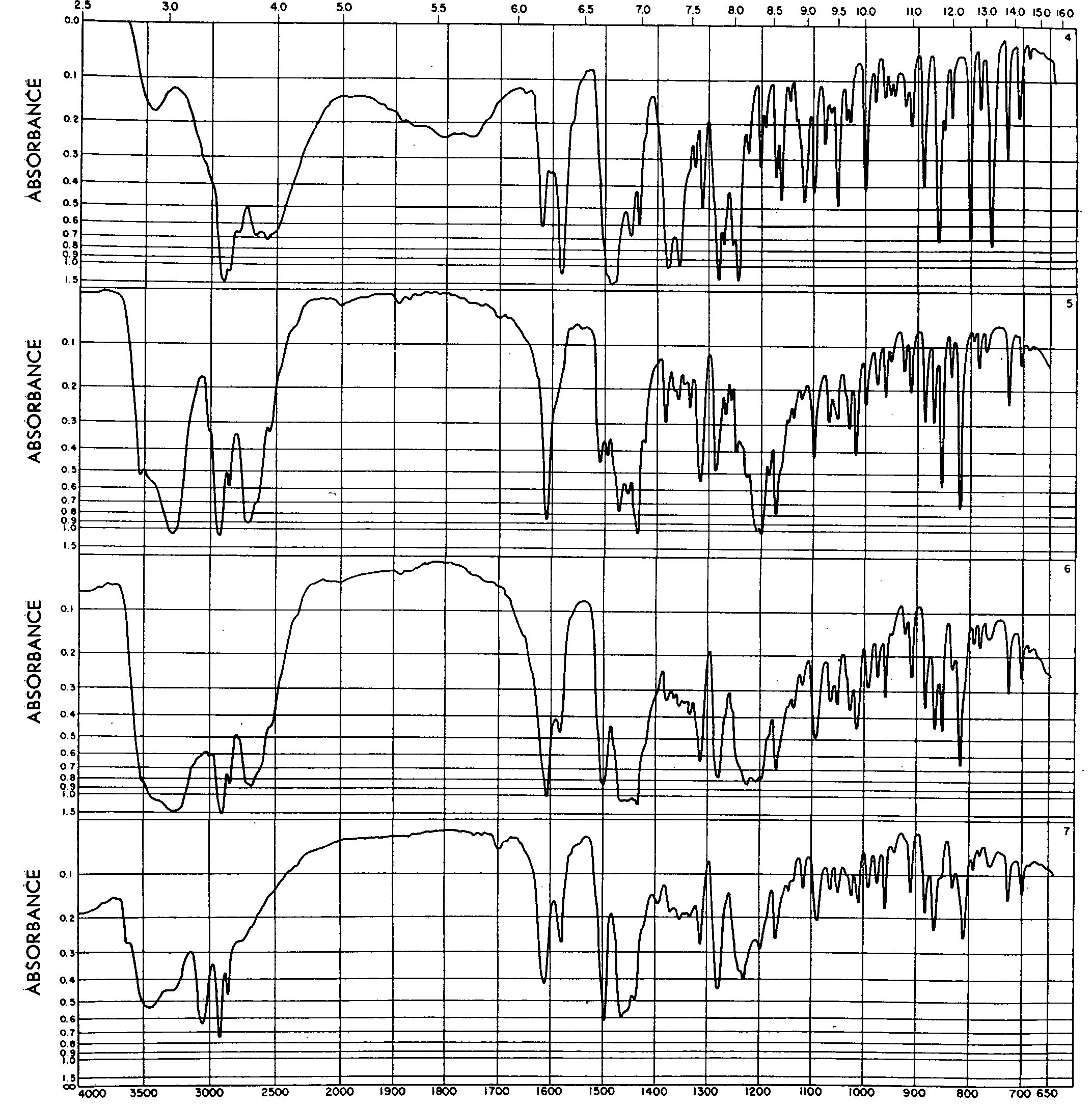
Infrared absorption spectra of the complex along with those of the narcotic-free base and its hydrobromide salt were obtained by the mineral oil mull and the pressed potassium bromide pellet technique. They were charted on a linear frequency Perkin Elmer Model 21 Double Beaω Recording Spectrophotometer, calibrated against atmospheric water-vapour and operated under the following conditions : slit auto; resolution 927; gain 5.9; response 1; source amperes 0.3; speed 3; suppression 0. The estimated errors in the frequencies reported are ± 5 cm-1throughout the 4000-2000 cm-1region and ± 2 cm-1beyond this wavelength range.
The mineral oil (Nujol) spectra were obtained by grinding finely with an electric vibrator a few mg of sample in the vehicle, placing the resultant smooth paste between two salt plates and measuring its absorption throughout the 4000-650 cm-1frequency range. A sodium chloride window was used in the reference beam during the experiments.
The KBr spectra were obtained by first powdering the specimens freely in a mechanical grinder until they would pass through a 250-mesh sieve (U.S. Standard Sieve Series No. 230). About 2 mg of sample were then mixed intimately with 400 mg of A.C.S. Reagent Grade potassium bromide prepared similarly and dried overnight at 125°C. A 200-mg aliquot of the mixture was subject in vacuo for about five minutes to a pressure of 10,000 lbs/sq. inch and the absorbance of the resulting clear disc recorded. A KBr window prepared under comparable conditions was placed in the path of the reference beam to compensate for absorption by the reagent.
Examination of the spectra reproduced in Figs. 7 and 8 shows that in marked contrast to the few (4-6) bands displayed in the ultraviolet, the compounds exhibit a great number of characteristic absorptions in the infrared. Furthermore, whereas all UV-characteristics of the narcotic (and its hydrobromide salt) are modified as a result of complex formation - see Fig. 3 - many of its prominent features are retained in the infrared. This special UV-IR interrelationship has inherent advantages and demonstrates the complementary nature of the two techniques.
Thus, in Nujol both the narcotic-free base and the complex show two intense bands (1618 cm-1and 1578 cm-1)none of which is present in the spectrum of the alkaloidal salt. Additional key bands appearing in the mineral oil spectra of the compounds are listed in Table VI. Similar common characteristics as well as differentiating features are observed in the KBr-spectra shown in Fig. 8. A more detailed interpretation of their significance in terms of structure is to make the subject of another paper supplementing the Atlas of Infrared Spectra of Narcotics and Related Alkaloids published by Levi, Hubley and Hinge [52a] .
|
Compounds | |||
|---|---|---|---|
|
Characteristic IR-absorptions (cm-l) |
Dromoran |
Dromoran. HBr |
[Dromoran.H] 2.[PtCl 6] |
| 3520 |
- |
weak |
strong |
| 3265 |
- |
strong |
- |
| 3225 |
- |
- |
strong |
| 3100 |
- |
- |
strong |
| 2700 |
- |
strong |
- |
| 1618 |
strong |
- |
strong |
| 1607 |
- |
strong |
- |
| 1578 |
strong |
- |
strong |
| 1505 |
- |
weak |
- |
| 1495 |
- |
strong |
strong |
| 1490 |
- |
strong |
- |
| 1375 |
strong |
weak |
strong |
| 1280 |
strong |
strong |
strong |
| 1240 |
strong |
- |
strong |
| 1170 |
medium |
strong |
medium |
| 1160 |
medium |
- |
weak |
| 1115 |
medium |
weak |
strong |
| 1095 |
medium |
strong |
- |
| 1090 |
- |
- |
strong |
| 1050 |
strong |
weak |
strong |
| 1000 |
strong |
- |
- |
| 910 |
weak |
medium |
medium |
| 885 |
strong |
strong |
strong |
| 865 |
- |
strong |
strong |
| 860 |
strong |
- |
- |
| 850 |
weak |
strong |
weak |
| 820 |
- |
strong |
- |
| 810 |
- |
- |
strong |
| 800 |
strong |
- |
- |
| 760 |
strong |
- |
weak |
| 730 |
medium |
- |
- |
| 725 |
- |
strong |
strong |
| 700 |
- |
weak |
medium |
| 695 |
medium |
- |
- |
Mechanism of the Reaction
The analytical data reported strongly suggest that the derivative is formed via interaction of the PtCl 6= ion, supplied by the reagent, with the alkaloidal cation, produced as a result of the virtually complete ionization of the salt in water.
1. H2PtCl6 ↔ 2 H+ +PtCl6
2. Dromoran H+ + Br-
3. 2 Dromoran H+ + PtCl6 ↔ [Dromoran H]2[PtCl6]
Further support for this mechanism of complex formation stems from the following experimental observations.
1. d,l-3-hydroxy-N-methylmorphinan (0.5 g) was dissolved in a solution of 50 ml of 1 N hydrochloric acid and 50 ml of ethanol. To this system was added slowly and with constant stirring a solution of 1 g of chloroplatinic acid in 50 ml of aqueous ethanol (1:1). The reaction product, isolated and purified in accordance with the procedure previously described, proved to be identical with that obtained when d,l-3-hydroxy-N-methylmorphinan hydrobromide was used as reactant.
2. d,l-3-hydroxy-N-methylmorphinan (0.5 g) was dissolved in 100 ml of 90% aqueous ethanol and to this solution were added slowly and with stirring 50 ml of an ethanolic 0.5% chloroplatinic acid solution. After refrigeration overnight the yellow precipitate was filtered off-because of the colloidal nature of the product this process was very slow-and dried in vacuo. Only 0.16 g of material was isolated which, after purification, proved to be identical with the preparation obtained by the previous method.
The decreased sensitivity of the process observed under these conditions is in accord with the proposed mechanism of the reaction. The alkaloidal base ionizes only partially in the solvent system used,
d,l- 3-hydroxy-N-methylmorphinan + H 2O
d,l- 3-hydroxy-N-methylmorphinan. H+ + OH
-hence the supply of cations is depressed and the reaction proceeds more sluggishly.
3. The reaction was carried out in dilute half-normal hydrobromic acid instead of half-normal hydrochloric acid. Again the product obtained showed the composition [Dromoran. H] 2.[PtCl 6]. All attempts to prepare a complex containing both bromine and chlorine ions - e.g., [Dromoran.HBr] 2. [PtCl] 4 or [Dromoran. HCl] 2. [PtBr 4] failed. These observations further support the suggested mechanism of the reaction in so far as they are in full accord with all the analytical data favouring the formula [Dromoran. H] 2. [PtCl 6] - rather than [Dromoran. HCl] 2. [PtCl 4] - for the isolated complex.
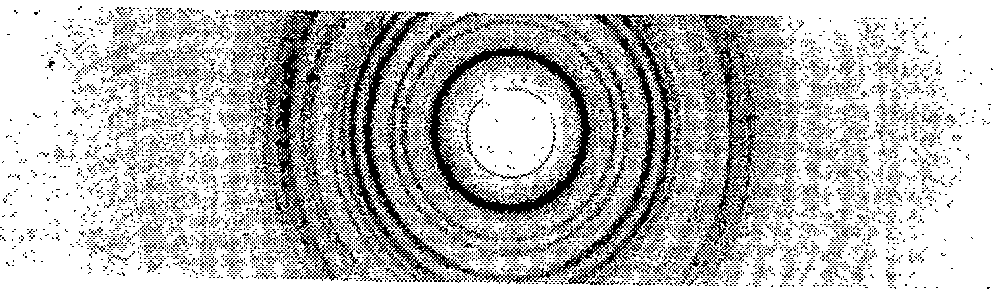
It was possible, however, to prepare the fully brominated compound by reacting the narcotic with platinic bromide in accordance with the following procedure. To a solution of 0.5209 g of d,l-3-hydroxy-N-methylmorphinan hydrobromide (0.0015 moles) in 50 ml of distilled water was added a solution of 0.7723 g of platinic bromide (0.0015 moles) in 50 ml of 1 N ethanolic hydrobromic acid. The precipitated complex was filtered off and dried over P 2O 5. (Weight of product = 0.860 g or 96.4% Theoretical Yield.) It was of a deep orange colour and following purification melted with decomposition from 249-253 °C. Quantitative analyses carried out in accordance with the procedure described earlier showed that it had the composition [Dromoran. H] 2 · [PtBr 6]. (Pt found: 16.04 %; Theoretical 16.37 %; Br found: 39.91% Theoretical 40.23%; Dromoran found: 42.69%; Theoretical 43.18%.) Its X-ray diffraction pattern is shown in Fig. 9 and its infrared absorption recorded in Figs. 7 and 8.


Microchemical Value of the Reaction
Under suitable conditions the reaction described becomes highly sensitive and may be used for the identification of trace amounts of the narcotic. The technique to be adopted for this purpose is as follows:
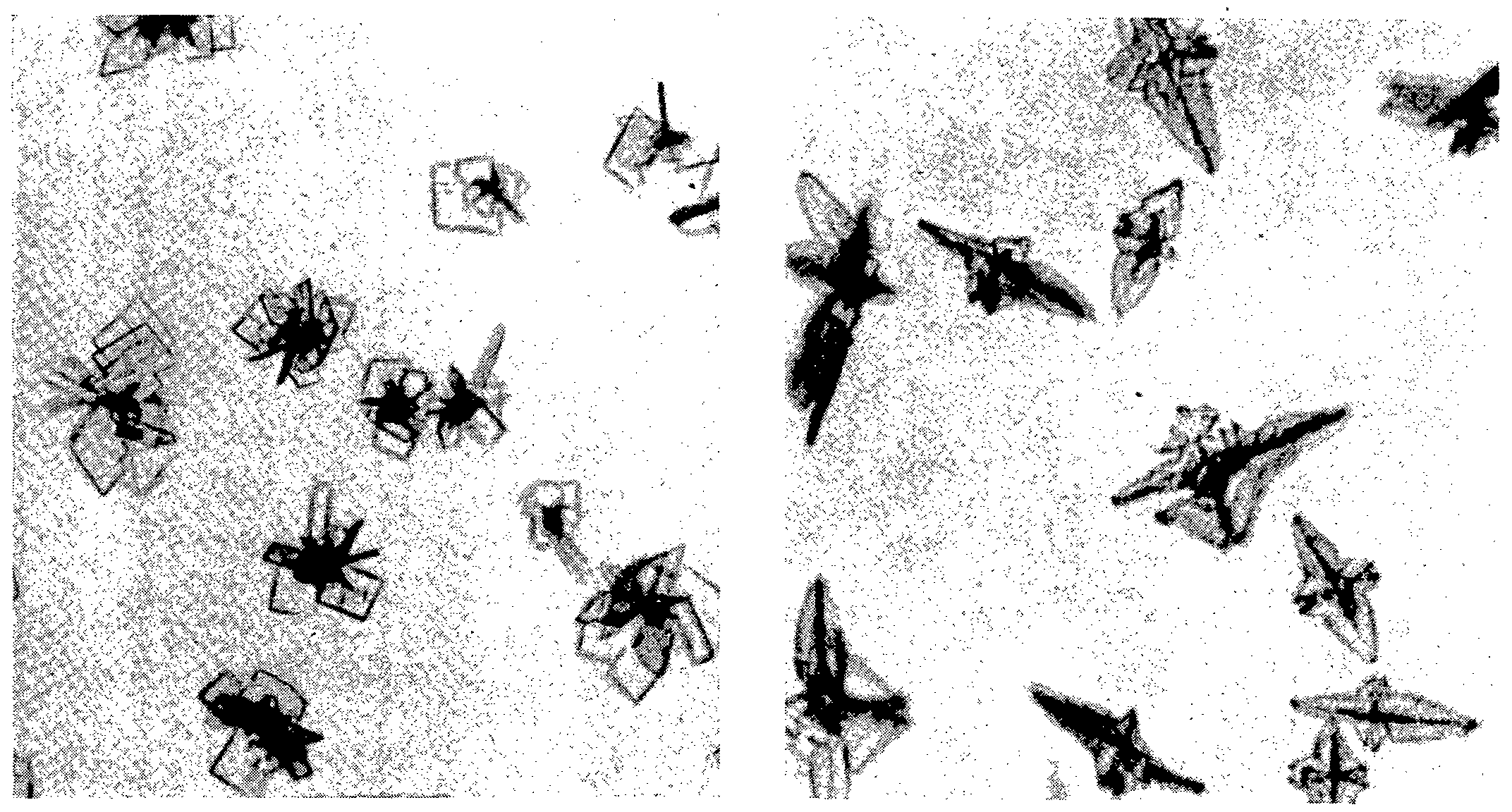
Weigh out 0.250 g of chloroplatinic acid and dissolve in a 100 ml of 50% ethanolic half-normal hydrochloric acid. Place a little of the specimen to be tested on a microscope slide and add a drop of the reagent. Immediately rosette-like crystals will form from the amorphous mass and start floating in all directions toward the edges of the test drop - see Figs. 10 and 11.
The reaction is particularly sensitive when carried out on the solid material. Hence, if a solution is to be analysed it is best to evaporate a drop of it to dryness on a microscope slide and perform the test directly on the residue. As little as 1 γ of the narcotic may thus be readily detected.


The sensitivity of the microchemical reaction described is matched by a high degree of specificity. No characteristic crystalline derivatives are obtained with morphine and morphine salts, ethylmorphine, dihydromorphine, dihydromorphinone, heroin hydrochloride, codeine phosphate, dihydrocodeine, dihydrocodeinone, metopon, thebaine, narcotine, Phenadoxone and Demerol. All of these drugs, with the exception of heroin hydrochloride, dissolve on addition of the reagent and only after evaporation of the ethanol from the slide will some of them start to crystallize from the medium. The reagent therefore exceeds by far the sensitivity and specificity of Reinecke's Salt, which yields micro-crystalline derivatives with the opium alkaloids as well [54] .
It should be pointed out, however, that the sensitivity of the test is not the same for all Dromoran compounds. The free base reacts somewhat more sluggishly than the hydrobromide salt and the tartrate is even less reactive than the free base. These observations further confirm the reaction mechanism described earlier, and accordingly the phenomenon may be overcome by adding to the sample on the microscope slide a drop of hydrochloric acid, evaporating the solution to dryness and performing the test on the residual salt.
The reagent may also be used for the microchemical identification of d- and l-3-methoxy-N-methylmorphinan* - see Fig. 12 - as well as of the d,l-isomer - see Fig 13. The observation that the latter compound yields different crystals than its optical antipodes is in line with the finding of Barnes and Sheppard [2] , who showed by X-ray analysis that the narcotic is not a mixture of two isomers but represents a true racemoid.
* Only l-3-methoxy-N-methylmorphinan is considered a narcotic. The d-isomer lacks analgesic properties [45] , [80] .
Additional Microchemical Identification Tests
Since this work was completed, several papers relating to the microchemical identification of Dromoran have appeared in the literature. Reference to these researches is made in a special section of the bibliography cited at the end of this article, so as to enable the forensic chemist and toxicologist to locate pertinent data more readily. Also, in private correspondence with Dr. Goldbaum, Chief, Division of Physiology and Pharmacology, Army Medical Service Graduate School, Walter Reed Army Medical Center, Washington, D.C., the author was told of a sensitive chromatographic method of alkaloid analysis applicable to Dromoran. Those interested in experimental details are advised to communicate with Dr. Goldbaum directly [35] .
The author is grateful to Dr. L. I. Pugsley for valuable advice and discussions, and to Mr. R. C. Hammond of the Division of Narcotic Control, for much helpful information. He also wishes to thank Dr. W. H. Barnes, of the National Research Council, for the X-ray diffraction patterns. Acknowledgement is also extended to Mr. R. Beaudry for technical assistance, to Mr. M. Butler for drafting the experimental results and to Messrs. E. C. Kerr and I. Dooh for preparing prints of the illustrative material.
BREINLICH, J., "Der Mikrochemische Nachweis einiger Analgetika, insbesondere Polamidon und Ticarda "Höchst ", Cliradon "Ciba " und Dromoran "Roche " , Arzneimittel Forschung, 3, 93 (1953).
"Nachweis von Dromoran und Cliradon neben Morphin im Urin durch Trübungsmethoden " , Arzneimittel Forschung, 3, 212 (1953).
"ZumMikrochemischen Nachweis von Cliradon und Dromoran ", Arzneimittel Forschung, 3, 490 (1953).
BROSSI, A., HAEFL1GER, O., SCHNIDER, O.: "Oxy-morphinane. 6. Die papierchromatographische Bestimmung von Morphinanderivaten und die Verfolgung ihrer Ausscheidung beim Hund ", Arzneimittel-Forschung, Feb. 1955, 5: 2, pp. 62-66.
GOLDBAUM, L. R., "TheApplication of Paper Chromatography in the Analysis of Alkaloids ".Private communication.
JATZKEWITZ, H., "Ein Klinisches Verfahren zur Bestimmung von Basischen Suchtmitteln in Harn ", Hoppe-Seyler's Zeitschr. Physiol. Chem., 292, 94 (1953).
KAISER, H., and JORI, H., "Beiträge zum Toxikologischen Nachweis von Dromoran ' Roche ', Morphin, Dilaudid, Cardiazol, Coramin und Atropin mit Hilfe der Papierchromatographie ", Pharmaz. Ztg -Nachr., 88, 963 (1952); Archiv der Pharmazie, 287, 224 (1954).
VIDIC, E., "Nachweis, Unterscheidung und Quantitative Bestimmung stark wirkender synthetischer Analgetika, insbesondere des Cliradon, im Urin ", Arzneimittel Forschung, 3, 34 (1953).
"Nachweis und Quantitative Bestimmung des Dromoran " , Arzneimittel Forschung, 3, 428 (1953).
"Zum Mikrochemischen Nachweis von Cliradon und Dromoran ", Arzneimittel Forschung, 3, 490 (1953).
BARLTROP, J. A., "Syntheses in the Morphine Series. Part. I. Derivatives of Bicyclo-(3, 3, 1)-2-Azanonane ", Jour. Chem. Soc., 399 (1947).
002BARNES, W. H., and SHEPPARD, H. M., "X-Ray Diffraction Powder Data for eighty three Narcotics ", Bull. Narcotics, 6 (2), (1954).
003BECKER, J., and BUNDSCHUH, O., Med. Klinik, 47, 546 (1952).
004BRACKETT, J. W., and BRADFORD, L. W., " Detection, Identification and Estimation of Non-volatile Organic Poisons, Drugs and Narcotics by Ultraviolet Spectrophotometry ". Paper presented at 123rd Meeting of the American Academy of Forensic Sciences, Los Angeles, Cal., March 1953.
005BRAENDEN, O. J., and WOLFF, P. O., "Synthetic substances with Morphine-Like Effect ", Bull. World Health Org., 10, 1003 (1954).
006BRAENDEN, O. J., EDDY, N. B., and HALBACH, H., "Synthetic Substances with Morphine-Like Effect. Relationship between Chemical Structure and Analgesic Action ". United Nations Economic and Social Council publication, E/CN 7/299, 30 March 1955.
007BRAND, J. C. D., HORNING, W. C., and THORNLEY, M. B., "The Spectrophotometric Determination of the Ionization Constants of Aromatic Nitro-Compounds ", Jour. Chem. Soc., 1374 (1952).
008BRAND, J. C. D., "Aromatic Sulphonation. The Ionization Constants of p-Nitrotoluene, Nitrobenzene and p-Chloronitrobenzene and the Acidity Function of Oleum ", Jour. Chem. Soc., 997 (1950).
009BRODE, W. R., "The Determination of Hydrogen Ion Concentration by a Spectrophotometric Method and the Absorption Spectra of Indicators ", Jour. Amer. Chem. Soc., 46, 581 (1924).
010BROTMAN, M., CULLEN, S. C., and WILKINS, D. S., "Intravenous Supplementation during Nitrous Oxide Anesthesia. Comparison of Demerol, Morphine and a New Potent Analgesic Drug ", Anesthesiology, 11, 527 (1950).
011BROWN, A. K., "Methorphinan as a Supplement to Nitrous Oxide and Oxygen Anesthesia ", Brit. Med. Jour., 1331 (1952).
012BRUSTIER, V., "Contributions à l'Etude spectrographique de l'absorption des rayons ultraviolets par les alcaloides et les glucosides ", Bull. Soc. Chim., 39, 1527 (1926).
"Ultraviolet Absorption Spectra of Alkaloids ", Chim. et Ind., 27, 1007 (1932).
013CLARK, W. A., and MCBAY, A. J., "Spectrophotometric Determination of Morphine and of Codeine ", Jour. Am. Pharm. Assoc., Sci. Ed., 43, 39 (1954).
014CRONYN, M. W.," Azabicycloalkanes. 2-Azabicyclo-(3, 3,1)-nonane" Journ. Org. Chem., 14, 1013 (1949).
015CURRERI, A. R., GALE, J. W., and DICKIE, H. A., "Effectiveness of Dromoran (3-Hydroxy-N-methylmorphinan) as an Analgesic in Thoracic Surgery ", Jour. Thoracic Surg., 20, 90 (1950).
016DEDE, L., and ROSENBERG, A.," über innere Molekülverbindungen ", Ber., 67B, 147 (1934).
017DOBBIE, J. J., LAUDER, A., and TINKLER, C. K., "The Constitution of Cotarnine ", Jour. Chem. Soc., 83, 598 (1903).
"The Relative Strengths of the Alkaline Hydroxides and of Ammonia as measured by their Action on Cotarnine ", Jour. Chem. Soc., 85, 121 (1904).
018DOBBIE, J. J., and LAUDER, A.," On the Relation between the Absorption Spectra and the Chemical Structure of Corydaline, Berberine and other Alkaloids ", Jour. Chem. Soc., 83, 605 (1903).
"The Absorption Spectra of Laudanine and Laudanosine in Relation to Their Constitution ", Jour. Chem. Soc., 83, 626 (1903).
"The Absorption Spectra of Cinchonine, Quinine, and Their Isomerides ", Jour. Chem. Soc., 99, 1253 (1911).
"The Absorption Spectra of Neopine ", Jour. Chem. Soc., 99, 34 (1911).
019DOBBIE, J. J., and FOX, J. J., "Relation between the Absorption Spectra and the Constitution of Certain Isoquinoline Alkaloids and the Alkaloids of Ipecacuanha ", Jour. Chem. Soc., 105, 1639 (1914).
020Document E/1998 issued by the Economic and Social Council, Department of Social Affairs, United Nations.
021Drug Submission by Hoffmann-LaRoche, Inc., Nutley, N.J., to the Department of National Health and Welfare, Ottawa, Canada, April 3, 1951.
022EDWARDS, L. J., "The Hydrolysis of Aspirin. A Determination of the Thermodynamic Dissociation Constant and a Study of the Reaction Kinetics by Ultraviolet Spectrophotometry ", Trans Farad. Soc., 46, 723 (1950).
023ELVIDGE, W. F., "Absorption Spectrophotometry in Pharmaceutical Analysis ", Quart, Journ. Pharm. Pharmac., 13, 219 (1940).
023a23a. FARMILO, C. G., "The Ultraviolet Spectrophotometric Method ", Bulletin on Narcotics, 6, 3-4 (1954).
024FISCHER, H., "Die Anwendung der Gerichtlichen Medizin als Fest-stellungsmethodik speziell zum Toxikologischen Nachweis von Alkaloiden ". Inaugural Dissertation zur Erlangung der Doktor-würde der Medizinischen Fakultät der Universität Zürich. Buch-druckerei Stäfa, A. G. vorm. E. Gull, Zürich, 1925.
"Die Physikalische Chemie in der Gerichtlichen Medizin und in der Toxikologie mit spezieller Berücksichtigung der Spektrographie und der Fluoreszenz Methoden ". Kommissionsverslag der Buchhandlung A. Rudolf, Zürich 1925.
025FLEXSER, L. A., HAMMETT, L. P., and DINGWALL, A., "The Determination of Ionization by Ultraviolet Spectrophotometry. Its Validity and its Application to the Measurement of the Strength of Very Weak Bases ", Jour. Am.. Chem. Soc., 57, 2103 (1935).
026FOSTER, C. E., and MACDONALD, J., "The Ultraviolet Spectrum of Papaverine Hydrochloride ", Jour. Pharm. Pharmac., 3, 127 (1951).
027FOX, J. J., and SHUGAR, D., "Absorption Spectra and Structure of Barbituric Acid Derivatives as a Function of pH ", Bull. Soc. Chim. Belg., 61, 44 (1952).
028FRASER, H. F., and ISBEL, H., "Addiction Liabilities of Morphinan, 6-Methyldihydromorphine and Dihydrocodeinone ", Jour. Pharmac. Exptl. Therap., 100, 128 (1950).
029FROMHERZ, K., Arch. Int. Phamacodyn., 85, 387 (1951).
030FULTON, C. C., "Alkaloids and Their Reagents ", Amer. Jour. Phar., 3, 183 (1939).
031GINSBURG, D., "The Synthesis of Morphan. 2-Azabicyclo-(3, 3, 1)-nonane ", Jour. Org. Chem., 15, 1003 (1950).
032GLAZEBROOK, A. J., "Actions and Uses of Methorphinan ", Brit. Med. Jour., 1328 (1952).
033GOLDBAUM, D. S., "A Spectrophotometric Determination of Isonicotinic Acid Hydrazide ", Science, 120, (No. 3112), 315 (1954).
034GOLDBAUM, L. R., "Determination of Barbiturates. Ultraviolet Spectrophotometric Method with Differentiation of several Barbiturates ", Anal. Chem., 24, 1604 (1952).
035GOLDBAUM, L.R., Private Communication. "The Application of Paper Chromatography in the Analysis of Alkaloids ", by Leo R. GOLDBAUM and Leo KAZYAK.
036GOLDBAUM, L. R.., and KAZYAK, L., " The Determination of Morphine in Urine using Paper Ionophoresis and Ultraviolet Spectrophotometric Analysis ", Jour. Pharmac., Exptl. Therap., 106, (4), (1952).
037GRADWOHL, R. B. H., "Legal Medicine ", The C. V. Mosby Co., St. Louis, Mo. (1954). Chapter 24, "Toxicology " by Sidney Kaye and Leo R. Goldbaum, pp. 599-723.
038GREWE, R., "Das Problem der Morphin-Synthese " Die Naturwissenschaften, 33, 333 (1946) (Translation to be found in United Nations Economic and Social Council publication, E/CN 7/154, 17 Feb. 1949).
039GREWE, R., "Synthesen in der Phenanthrene-Reihe ", Berichte, 72, 426, 785, 1314 (1939); 76, 1072, 1076 (1943).
039aGREWE, R., "SynthetischeArzneimittel mit Morphin-Wirkung ", Angewandte Chemie, 59, 194 (1947).
040GREWE, R., and Mondon, A., "Synthese des Morphinans ", Berichte, 81,279 (1948).
041GRUBER, Jr., C. M., Soo LEE, K., and GRUBER, C. M.," A Comparison of the Actions of Meperidine, NU-1196, Methadone, Morphine, NU-2206 and Metapon upon the Intestine and Uterus ", Jour. Pharmac. Exptl. Therap. , 99, 317 (1950).
041aGULLAND, J. M. and ROBINSON, R., "Constitutionof Codeine and Thebaine ", Mem. Proc. Manchester Lit. Phil. Soc., 69, 79 (1955).
042HAGEMEYER, G., Mediz. Welt, 20, 1286 (1951).
043HARNASCH, H., Münch. Mediz. Wschr. (4), 164 (1952).
044HARTLEY, W. N., "The Absorption Spectra of Alkaloids ", Phil. Trans. Roy. Soc., London, 176, 471, 516 (1885).
045ISBELL, H., and FRASER, H. F., "Human Pharmacology and Addiction Liability of Derivatives of 3-Hydroxy-N-methylmorphinan ", Jour. Pharmac. Exptl. Therap., 103, 348 (1951).
"Actions and Addiction Liabilities of Dromoran Derivatives in Man ", Jour. Pharmac. Exptl. Therap., 107, 524 (1953).
046JACKSON, G. R., WESCHLER, J. R., and DANNLEY, R. L., "Ultraviolet Absorption Spectra of the Hydrolysis Products of Diethyl barbituric Acid ", Anal. Chem., 26, 1661 (1954).
047JAGGARD, R. S., ZAGER, L. L., and WILKINS, D. S.," Clinical Evaluation of Analgesic Drugs. A Comparison of NU-2206 and Morphine Sulfate administered to Postoperative Patients ", Arch. Surg., 61, 1073 (1950).
048JUNKERMAN, C. L., HEEN, R. C., and POHLE, H. W.," Clinical Experience with a New Analgesic Agent ", Jour. Lab. Clin. Med., 34,1615 (1949).
049KATO, S., and SOMENO, F., "Absorption Spectrum and Molecular Structure ", Sci. Papers Inst. Phys. Chem. Research (Tokyo), 33,209 (1937), 34,905 (1938).
050KEUTMANN, E., and FOLDES, F. F., "The Analgesic Effect of Dromoran Hydrobromide in Postoperative Pain ", New England Jour. Med., 244, 286 (1951).
051KITASATO, Z., "Beiträge zur Kenntnis der Isochinolin Alkaloide ", Acta Phytochimica, Vol. 2 (No. 2), 175 (1927).
052KUMLER, W. D., and STRAIT, L. A., "The Ultraviolet Absorption Spectra and Resonance in Benzene Derivatives ", Jour. Amer. Chem. Soc., 65, 2349 (1943).
052aLEVI, L. ,HUBLEY, C. and HINGE, R., "Infrared Spectra of Narcotics and Related Alkaloids ", Bull. Narcotics, 7, 1 (1955).
053LEVI, L., and FARMILO, C. G., " Reaction of Morphine with Potassium Mercuric Iodide ", Anal. Chem., 26, 1040 (1954).
054LEVI, L., and FARMILO, C. G., "The Characterization of Narcotics as Reineckates ", Can.Jour. Chem., 30, 783 (1952).
055LOOFBOUROW, J. R., and STIMSON, M. M., "Ultraviolet Absorption Spectra of Nitrogenous Heterocyclic Compounds ", .Jour. Chem. Soc., 1275 (1940).
056LOTHIAN, G. F.," Absorption Spectrophotometry ", Hilger and Watts, Ltd., London, 1949.
057MORGAN, Ch. E., and CEPRINI, M. Q., "Buffered Drug Solutions for Measurement of Ultraviolet Absorption at various pH's ". Reprints of this paper may be obtained from Ch. E. Morgan, Chief Chemist, State of New York, Department of State, Racing Commission Laboratory, 148-11 Hillside Ave., Jamaica 35, N.Y., U.S.A.
058MORTON, R. A., and TIPPING, A. H., "A Spectroscopic Method for the Determination of Dissociation Constants ", Jour. Chem. Soc., 1398 (1926).
059M?HLAY, M.,ärztl . Wschr., 7, 257 (1952).
060OSOL, A., "The Spectrophotometric Determination of the Dissociation Constants of Theophylline, Theobromine and Caffeine ", Jour. Am. Pharm. Assoc., Sci. Ed., 38,158 (1949).
061OESTREICHER, P. M., FARMILO, C. G., and LEVI, L.,"Ultraviolet Spectral Data for Ninety Narcotics and Related Compounds ", Bull. Narcotics, 6, 3-4 (1954).
062PRINGSHEIM, P., "Fluoreszenzund Phosporeszenz im Lichte der Neuen Atom Theorien ", J. Springer Verlag, Berlin, 1921.
063RANDALL, L. O., and LEHMANN, G., "Analgesic Action of 3-Hydroxy-N-methylmorphinan Hydrobromide (Dromoran)", Jour. Pharmac. Exptl. Therap., 99, 163 (1950).
064RECHENBERG, H. K., and ZEERLEDER, R., Schweiz. Med. Wschr., 81, 44 ,1086 (1951).
065SCHNIDER, O., and GRUSSNER, A.," Synthese von Oxy-morphinanen ", Helv. Chim.. Acta, 32, 821 (1949).
066SCHINDER, O., and GRUSSNER, A., "Oxy-morphinane. (3. Mitteilung).Optisch aktive 3-Oxy-morphinane ", Helv. Chim. Acta, 34, 2211(1951).
066aSCHINDER, O., and HELLERBACH, J., "Synthese Von Morphinanen " (2. Mitteilung), Helv. Chim.. Acta, 33, 1437 (1950).
"Oxymorphinane (4. Mitteilung); Synthese Von Benzyl-octahydroisochinolinen ", Helv. Chim. Acta, 34, 2218 (1951).
066bPaper presented by Dr. E. L. SEVRINGHAUS, Vice-President for Chemical Research, Hoffmann-LaRoche, Inc., Nutley, N.J., before Officers of the Food and Drug Directorate and the Division of Narcotic Control, Ottawa, Feb. 14, 1951.
067SIEGEL, B. M., BLOCH, D. P., PITESKY, I., and LAST, J. H., "Comparative Study of Morphine and Dromoran as Antidiuretic Agents in the Dog ", Proc. Soc. Exptl. Biol. ,Med., 74, 809 (1950).
068SLOMKA, M. B., and GROSS, E. G., "An Evaluation of the Analgesic Activity of the Dromoran Isomers ", Proc. Soc. Exptl. Biol. ,Med., 81,548 (1952).
069SMITH, H. W., and MACDOUGAL, J. R., "A Compilation of Spectrophotometric Data on Compounds of Toxicological Interest ". Attorney General's Laboratory, Toronto, Canada, May 1953.
070SMITH, R. M., "Clinical Experience with Dromoran Hydrobromide (NU-2206) for the Relief of Chronic Pain ", South. Med. Surg., 112, 251 (1950).
071STENSTRÖM, W., and GOLDSMITH, N., "Determination of Dissociation Constants of Phenol and the Hydroxyl Group of Tyrosine by means of Absorption Measurements in the Ultra-Violet ", Jour. Phys. Chem., 30, 1683 (1926).
072STENSTROM, W., and REINHARD, M., "The Influence of the pH upon the Ultraviolet Absorption Spectra of Certain Cyclic Compounds ", .Jour. Phys. Chem., 29, 1477 (1925).
073STIMSON, M. M., and REUTER, M. A., "Spectrophotometric Estimation of Methoxycinchona Alkaloids ", Jour. Am. Chem. Soc., 68, 1192 (1946).
074STOELTING, V. K., THEYE, R. A., and GRAF, J. P., Anesthesiology,12,225 (1951).
075STUCKEY, R. E., "The Ultraviolet Absorption Spectra of Barbituric Acid and its 1-Methyl and 1, 3-Dimethyl Derivatives ", Quart. Jour. Pharm. Pharmac., 13, 312 (1940).
"The Absorption Spectra of Organic Compounds containing Nitrogen. Part. I. Derivatives of Hydantoin ", Jour. Chem. Soc., 331 (1947). "The Applications of Ultraviolet Absorption Spectrophotometry in Pharmaceutical Analysis ", Jour. Pharm. Pharmac., 4, 345 (1952).
076TREADWELL, F. P., and HALL, W. T., "AnalyticalChemistry ". John Wiley & Sons, Inc., New York, 1952. VoL I, pp. 281, 321, 522.
077VANDENBELT, J. M., and DOUB, L., "The Ultraviolet Absorption Spectra of Simple, Unsaturated Compounds ", .Jour. Am. Chem. Soc., 69, 2714 (1947).
078World Health Organization, Expert Committee on Drugs liable to produce Addiction, Technical Report Series, No. 21, March 1950, pp. 6 and 11. Note submitted by Dr. N. B. EDDY, Medical Officer, National Institutes of Health (U.S. Public Health Service), Bethesda, Md., U.S.A., based upon the work of Dr. H. ISBELL, Research Director, U.S. Public Health Services Hospital, Lexington, Ky., U.S.A.
079World Health Organization, Expert Committee on Drugs liable to produce Addiction, Technical Report Series, No. 57, January 1952, pp. 6 and 7.
080World Health Organization, Expert Committee on Drugs liable to produce Addiction, Technical Report Series, No. 76, March 1954, pp. 8 and 9.
081WILKINS, D. S., CULLEN, S. C., and BROTMAN, M., Anesthesiology 12, 145 (1951).
082WOHL, A., "étude spectrale sur les arylamines et leurs chlorhydrates ", Bull. Soc. Chim. France, 6, 1312 (1939).
"L'absorptiondans l'ultraviolet des sels de Diazonium ", Bull. Soc. Chim. France, 6, 1319 (1939).
083ZAGER, L. L., SAWTELLE, W. W., GROSS, E. G., NAGYFY, S. F., and TIDRICK, R.T., "Observations on the Use of a New Analgesic, NU-2206 (3-Hydroxy-N-methylmorphinan Hydrobromide)", Jour. Lab. Clin. Med., 34, 1530 (1949).

