Physico-chemical methods have recently been gaining in importance as a means of analysing drugs, particularly alkaloids. Yet although polarography is now a standard procedure in all analytical laboratories, relatively little use is so far being made of oscillographic polarography. One reason for this is that it is a new method which-at least with respect to its general use for analytical purposes-is still in the initial stages of its development.
Author: Robert Kalvoda, Jaroslav Zyka
Pages: 41 to 45
Creation Date: 1957/01/01
Physico-chemical methods have recently been gaining in importance as a means of analysing drugs, particularly alkaloids. Yet although polarography is now a standard procedure in all analytical laboratories, relatively little use is so far being made of oscillographic polarography. One reason for this is that it is a new method which-at least with respect to its general use for analytical purposes-is still in the initial stages of its development.
Since we believe that extensive use could be made of oscillographic polarography for purposes of narcotics analysis, we propose in this report to discuss our experience with this method-in relation to compounds and narcotics problems-and at the same time to present a few examples of the experimental results we have recently obtained. An introduction is devoted to a description of the method itself and the apparatus required.
Introduction
Oscillopolarography is the study of electrolysis using polarizable mercury electrodes connected to an oscillograph, on whose screen various relations between voltage, current intensity and time may be recorded.
Depending on which of these functions is being observed, oscillopolarographic methods fall into two categories, according as the polarizable electrode is polarized by voltage or by current. The second method, developed and applied by Heyrovsky (1), (2), was at first used mainly to supplement the information available on polarographic phenomena, since it was possible by this means to observe these phenomena at mercury capillary electrodes under extremely rapid changes in potential. It emerged in the course of this work that there would also be great advantages in using the same method for analytical purposes.
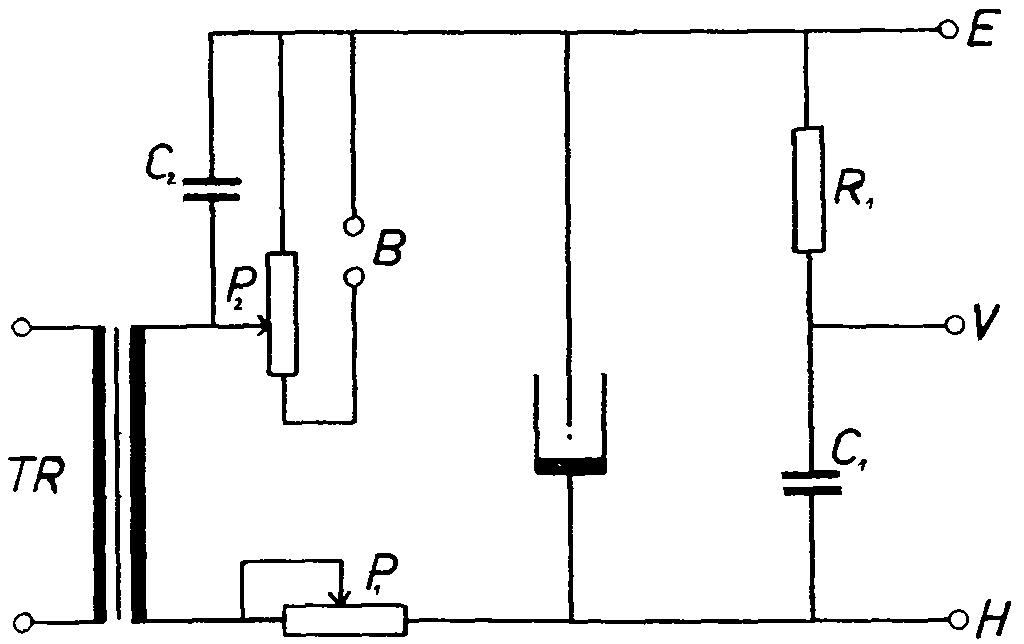
FIG. 1. - Diagram of Apparatus connected for Observation of dV/dt = f/V Curves
P 1 = Potentiometer 0.5 MΩ, P 2 = Potentiometer 5 KΩ, B = Battery 50-100 V, C 1 = Condenser 30,000 pF, C 2 = Condenser 2 μF, R 1 = 1,000 Ω, TR = Transformer 220/150V, E = to earth terminal of oscillograph, H = to horizontal amplifier of oscillograph, V = to vertical amplifier of the oscillograph.
In oscillographic polarography with alternating current the following functions apply:
V = f(t) - curves given by the time/potential function of the polarizable electrode under constant alternating current.
dV/dt = f(t) - curves given by the magnitude of the time/potential changes as a function of time.
dV/dt = f(V) - curves given by the magnitude of the time/potential changes as a function of the potential.
In the experiments described below the function dV/dt = f(V) was used.
The oscillogram of this function for an "empty" electrolyte is elliptical in shape, the upper part corresponding to the cathodic, and the lower part to the anodic direction of the current passing through the capillary electrode. In the presence of a depolarizer, inflexions appear on both parts, their size being a function of the concentration. The position of the inflexion on the potential axis indicates the potential, which is closely related to the half-wave potential in conventional polarography; the depolarizer can therefore be determined qualitatively from the position of the inflexion. The relation between the positions of the cathodic and anodic inflexions is an indication of reversibility; where the inflexions lie opposite one another the reaction is reversible, and where they are apart, or if one of the inflexions fails to materialize, it is irreversible.
The apparatus used for recording these curves is very simple; it can consist of a standard commercial direct-current oscillograph with a built-in horizontal and vertical amplifier connected to a Heyrovsky polarograph. In this kind of apparatus (fig. 1) a suitable current intensity is applied by means of resistance P (0.3 - 1 mA for a dropping mercury electrode and up to 10 mA for a streaming electrode); a direct-current component is applied by means of resistance P in such a way that both lateral points on the ellipse are visible. The first point represents the potential at which the mercury is oxydized, and the second the potential at which the cation of the basic electrolyte is reduced.
A special electronic Polaroscope (3) has recently been constructed (by the Kovo Company, Prague VII), in which the entire apparatus is self-contained, so that all that need be done is to attach the cathode and the anode to the terminals and, after switching on, to apply a suitable current intensity and a direct-current component. The sharpness, brightness Polaroscope and size of the image can be regulated by adjusting the other controls (fig. 2). Once these simple operations have been carried out, which takes only a few seconds, the results of experiments may be observed directly on the screen. As regards the electrodes, both dropping mercury electrode and streaming mercury electrodes are used in practice. For most determinations, however, the use of a dropping mercury electrode and a mercury pool as the unpolarizable (reference) electrode is quite adequate. For small-scale work a graphite electrode may be used as the reference electrode (anode) (6). As the circuit can be closed simply by immersing the two electrodes in the solution, special vessels are unnecessary for routine determinations in air. This method also offers the further advantage that only a small amount of solution is required for making oscillopolarographic records. We have even made reliable analyses using about 0.5 ml of the test liquid.

When a dropping mercury electrode is used, a trace of changing size is observed on the oscilloscope screen. The trace is largest when the drop falls, and its size diminishes as the surface area of the drop increases (if a streaming mercury electrode is used the size of the trace remains unchanged).
For the purpose of recording the results obtained, the traces are photographed; if a dropping mercury electrode is used, the trace on the screen of the apparatus is usually photographed before the drop of mercury falls. The exposure given is about 1/25 sec., at maximum aperture. Agfa Isopan, Agfa Fluorapid and similar films are the most suitable; development is carried out in a hard working developer.
Oscillographic curves may also be photographed with the use of dropping mercury electrodes in such a way that the exposure is made as the drop "breaks away" and a new drop begins to form. Using an exposure of ? to one second, we obtain a trace on which, in the area of the image interwoven with the contour-lines of the individual cycles, there remains a dark tract, the outline of which is the oscillogram of the last cycle before the drop fell. If a regular dropping time is maintained, these images can also be used for quantitative determinations, by means of a calibration curve, the distance between the apex of the inflexion and the potential axis to the second power of the concentration being plotted against one axis.
Since the currents passing through the vessel in which the determination is carried out are greater in oscillographic polarography than in classical polarography, a more concentrated supporting electrolyte, about 1 M, must be used, unless special equipment is employed to compensate for the potential drop in the solution.
As has already been stated, the position of the inflexion-in relation to the potential axis-indicates the quality of the depolarizer. On the other hand, the depth of the inflexion is a function of the concentration. These two functions can be used for purposes of identification; for quantitative purposes and evaluations use may be made of some of the methods described below.
Comparative Titration
In this method, two curves of the function dV/dt = f(V) appear on the oscillograph screen, one of them corresponding to the test solution and the other to the control solution, which is titrated with a measured solution of a known depolarizer until both curves (or the depths of the inflexions) coincide. The amount of depolarizer in the known solution is determined from the amount of solution used up (5), (6).
In this case two dropping mercury electrodes are used (one for the test solution and the other for the control solution), made from capillaries of the same shape, 0.08 mm in diameter. A regular mercury drop rate (synchronized dropping) is ensured by the use of a mechanical vibrator. For the purpose of connecting the two electrodes alternately, a relay switch, operating at a rate of 25 switches per second, may be used. Measurements with this method are carried out as follows: an equal amount of indifferent electrolyte is first pipetted into the two electrolytic vessels and a suitable polarizing current intensity applied. In addition, a direct current component is applied simultaneously to both units. The test solution is then added to the supporting electrolyte in one of the vessels. The supporting electrolyte in the other vessel is titrated, using a microburette, with a measured solution of the known depolarizer, dissolved in the supporting electrolyte. Titration is continued until both traces on the screeen overlap. If both curves coincide, the concentration of the two solutions must be the same.
A sample photograph illustrating this type of comparative titration is given in figure 3.
Measuring the Depth of the Inflexions
As has already been stated, the depth of the inflexion, within a given concentration range, is a function of the concentration of the depolarizer. During the reduction processes these inflexions make their appearance from depolarizer concentrations of 0.5 M - 1.10-4 M. Their depth may be measured by a simple apparatus which projects into the oscillograph sreen a horizontal axis, adjustable vertically by means of a calibrated potentiometer.

FIG. 3. - Comparative titration
One curve represents the sample under analysis, of volume Pb, the other the basic electrolyte, which is titrated with a measured solution of a known depolarizer (Pb).
Rapid determination of depolarizers can be effected with this apparatus by means of a calibration curve. At each concentration of the depolarizer corresponding to a point on the calibration curve we adjust the projected axis by means of a calibrated shunt so that it is just touched by the apex of the inflexion on the oscillogram curve immediately before the drop falls. The appropriate value indicating the distance between the apex of the inflexion and the potential axis is read off on the shunt scale and plotted on a graph against the concentration. In the method based on measuring the depth of the inflexion, a separate calibration curve must be prepared for each depolarizer and the determination of the test sample carried out under precisely the same conditions as the calculation of the calibration curves, particularly as regards the value of the polarizing current, the direct-current component and the dropping time. This method is particularly suitable for serial control determinations, for example, of impurities in samples, in which case the horizontal axis is adjusted according to the inflexion of the permissible amount of impurity. Samples containing larger amounts of impurities are identified from the fact that the inflexions extend in depth beyond the horizontal axis. Samples must in all cases be suitably diluted to ensure that the depolarizer concentration varies within the range in which the inflexions can be read most easily, i.e. about 10-3 to 10-4 M. Errors with this method of measurement rarely exceed ± 3 per cent. An oscillographic curve with a horizontal axis is shown in figure 4.
Oscillographic Titration
In oscillographic titration the depolarizer is titrated with a solution which reduces its concentration, by complex formation or precipitation for example. The point of equivalence is reached at the moment of disappearance of the depolarizer inflexion. As an example we may cite the determination of indium and cadmium where both are present together by titration with complexing agent No. III (7).
Another method has been described in which the depo1arizer solution is diluted with the supporting electrolyte until the inflexion of the depolarizer disappears (8).
Oscillographic polarography has so far been used most widely in qualitative experiments and purity tests in connexion with the control of drugs, mainly alkaloids. For a clear description of this method we refer the recorder to the work of Heyrovsky (2), (9), (10), Kalvoda (11-13), and Dolezal (14).
Alkaloids
The practical application of oscillographic polarography was shown to be feasible by Kalvoda (11-13), who studied the behaviour of papaverine, pilocarpine, codeine, morphine, ethylmorphine, physostygmine, ephedrine, santonin, quinine and quinidine. The behaviour of physostygmine (eserine) is also of interest because two inflexions appear in a 1 M - KOH medium; however, after about one minute one of them disappears and at the same time the solution turns red, since rubreserine is produced. Molnar (15) and Parrak (16) have studied the behaviour of the alkaloids of quinine.

FIG. 4. - Oscillographic Curve with a Horizontal Axis, adjustable in the vertical plane
Research has also been carried out on the possibility of determining marceine, cotarnine, hydrastine and emetine (17), (18), and of identifying atropine and eumydrine in a mixture and certain other alkaloids (20), (21).
Dusinsky (22), (23) has studied the oscillopolarographic behaviour of morphine, apomorphine and lobeline.
Nicotine and trigonelline can be determined, as has been shown by Volke and Volkova (24), even in the presence of acids of nicotine.
Habersberger and Zyka (4) have made a detailed study of tropane and piperidine alkaloids (cocaine, tropacocaine, atropine, apoatropine, homatropine, pelletierine, coniine, pseudopelletierine and lobeline).
In conclusion, we may sum up the information so far acquired from the study of the oscillopolarographic behaviour of alkaloids as follows. Most alkaloids have a depolarizing effect in an alkaline hydroxide medium, but it cannot be assumed that this is due to the direct reduction of the test substances. The inflexions produced in this way are observed even with very low concentrations of alkaloids (about 10-5 M and less), they have various shapes, and often exhibit a time function. In general their potential is about -1.5 to -1.6 V; only in certain cases, therefore, can they be identified in mixtures.
Narcotics
Some of the alkaloids mentioned in the preceding section are compounds belonging to the group of substances having narcotic effects. Since we feel that the oscillopolarographic determination of such compounds might be useful, we shall discuss them in more detail.
Morphine (22), for example, cannot by itself be identified by the oscillopolarographic method; however, it can be converted into oxy-dimorphine, which produces the characteristic (reversible) inflexion at about -0.65 V. The reaction is produced by adding a solution of potassium ferricyanide (0.03 ml 0.5 N - K 3FeCN 6 to 20 ml) to a morphine solution in 1.25 N - NaOH. In concentrations of morphine higher than 2.10-4 M additional barely visible inflexions appear at the most negative potentials. Of the other opium alkaloids studied, this reaction is produced only by diacetylmorphine which hydrolyzes in an alkaline solution and reacts in the same way.
 FIG. 5. - Determination of morphine and papaverine in a mixture, in a 1.25 N - NaOH medium
FIG. 5. - Determination of morphine and papaverine in a mixture, in a 1.25 N - NaOH medium
The first inflexion from the left is produced by the morphine, which has first been oxidized with ferricyanide; the second inflexion on the cathodic curve indicates the presence of papaverine (according to Dusinsky (22).
Papaverine in 1.25 N - NaOH produces an inflexion at -1.65 V (in papaverine concentrations of over 8.10-5 M another inflexion appears at -1.75 V), and it can also be distinguished from other alkaloids of opium and determined in various drug forms; for example, papaverine was determined in Ipecosit-Spofa injections and in artificially prepared mixtures in the presence of thebaine, codeine, morphine, narcotine, ethylmorphine and codeine. Figure 5 demonstrates that morphine and papaverine can be determined simultaneously where both are present together.
The same method also permits the determination of 2 mg of morphine in a 10-ml sample of widely different substances; it does not, for example, destroy tinctures of vegetable extracts and other ballast materials. Organic solvents must first be removed by distillation.
The behaviour of apomorphine in a 1.25 N - NaOH medium has also been studied; at first four inflexions appear on the oscillographic curve, at potentials of -0.60 V, -0.75 V, -0.85 V and -1.10V. In concentrations higher than 1.10-4 M another inflexion appears, at -0.3 V. After some time a phenomenon typical of apomorphine occurs; the inflexion at -0.60 V disappears after 15 to 45 minutes and simultaneously the inflexion at -0.85 V grows deeper. These changes, which can be seen taking place on the oscillographic curve, are accompanied by a change in colour from yellow-red to green. They occur very rapidly if the sample is heated to about 80°, and are obviously caused by the disintegration of the molecules due to the severance of the bonds between the =CO groups.
Careful research has also been carried out on the oscillopolarographic behaviour of cocaine in various media (4), from acid to strongly basic types. Good results have been obtained using as media solutions of neutral salts, in which chloride of cocaine in concentrations of about 10-3 M produces two inflexions on the cathodic and anodic sides, the most negative being sharply defined even in cocaine concentrations of 5.10-5 M.
Very clearly defined inflexions are produced by cocaine in a hydroxide medium (KOH, LiOH, NaOH); these inflexions, however, diminish with time and eventually disappear. In a cocaine concentration of 5.10-4 M for example, the inflexion disappeared after about 4 to 5 minutes.
For cocaine concentration from 1.10-3 M to 5.10-5 M in a 0.8 M - KOH medium, inflexions appear at a potential of -l.4 V vs. the saturated calomel electrode (fig. 6).
In a 5.5 M - NaOH medium, using approximately the same depolarizer concentrations, an inflexion appears at a potential of -1.25 V vs. the saturated calomel electrode (fig. 7).
With this medium, in which, after mixing, a visible turbulence is already observable in the test solution, a corresponding anodic inflexion is also clearly visible.
For analytical purposes, such as the detection of cocaine, the most suitable medium in our opinion is a hydroxide solution, which produces very clear inflexions. When this medium is used determinations can also be made, with the aid of a calibration graph, on the basis of the functional relationship between the depth of the inflexion and the cocaine concentration. Also of interest is the possibility of using, for semi-quantitative purposes, the phenomenom described above, namely, that the depth of the inflexion decreases with time. This can be done merely by measuring the depth of the inflexion at a certain moment after the sample solution has been added to the supporting electrolyte and stirred, or by calculating the time taken for the tip of the inflexion to reach a certain horizontal level in relation to the potential axis, this time being to a considerable extent the linear function of the alkaloid concentration.
No generally valid explanation can yet be given for the oscillopolarographic behaviour of cocaine and other not directly reducible alkaloids, and in particular for their behaviour in solutions of highly concentrated hydroxides, in which the inflexions produced by certain alkaloids are extraordinarily sharp. In one of the most recent works on the subject (4), it has been suggested that this might be due to the depolarizing effect produced by suspensions which may, in strong hydroxide solutions, result (in the case of cocaine, for example) in the precipitation of the alkaloid base (25).

FIG. 6. - A 10-4 M solution of cocaine chloride in a 0.8 M - KOH medium, twenty seconds after mixing
Conclusion
We have attempted in this paper to outline briefly the technique of oscillographic polarography by means of alternating current, and to indicate its importance and possible use in the identification and purity testing of substances. We have described work carried out on the oscillopolarographic behaviour of alkaloids, with particular reference to morphine and cocaine.
We consider that in this particular field, the determination of narcotics, oscillographic polarography could be more widely used. It is a rapid method, and once the apparatus has been connected a determination can usually be made in a matter of a few seconds. Precisely because of its speed, increasing use is being made of the method in widely different branches of chemical analysis. Oscillopolarographic analysis is still at an early stage of development; however, it is now making very rapid progress, and there is every likelihood that the quantitative technique too will be extensively applied in the near future.
We have given a few examples of the possibilities offered by this method. There is every reason to believe that narcotics analysis is one of the fields in which oscillopolarography opens up new lines of research.
FIG. 7. - A 10-4 M solution of cocaine chloride in a 5.5 M - NaOH medium, fifteen seconds after mixing
HEYROVSKY, J., FOREJT, J. : Oscilografické polarografie, State Technical Literature Publishing House, Prague, 1953.
HEYROVSKY, J. : Collection, 18, 749 (1953).
HEYROVSKY, J. : Anal. Chim. Acta, 12, 600 (1955).
HABERSBERGER, K., ZYKA, J. : Českoslov. farm., 5, 264 (1956).
KALVODA, R., MACKU, J. : Collection, 20, 254 (1955).
KALVODA, R. : Chem. listy, 49, 759 (1955).
TREINDL, L. : Chem. listy (printing).
MOLNAR, L. : Chem. Zvesti, 8, 912 (1954).
HEYROVSKY, J. : Anal. Chim. Acta, 8, 283 (1953).
HEYROVSKY, J. : Českoslov. farm., 2, 403 (1953).
KALVODA, R. : Českoslov, farm., 3, 124 (1954).
KALVODA, R. : Českoslov. farm., 4, 501 (1955).
KALVODA, R. : Pharmazie, 11, 101 (1956).
DOLEZAL, J. : Chemie (printing).
MOLNAR, L. : Acta Chim. Hung. (printing).
PARRAK, V. : Českoslov. farm., 4, 337 (1955).
PARRAK, V. : Českoslov. farm., 4, 504 (1955).
PARRAK, V. : Pharmazie, 2, 205 (1956).
PARRAK, V. : Českoslov. farm., 4, 180 (1955).
PARRAK, V. : Českoslov. farm., 5, 84 (1956).
PARRAK, V. : Pharmazie, 11, 591 (1956).
DUSINSKY, G. : Českoslov. farm., 4, 400 (1955).
DUSINSKY, G. : Pharmazie, 11, 42 (1956).
VOLKE, J., VOLKOVA, V. : Chem. listy, 48, 1031 (1954).
MICKA, K. : Chem. listy, 49, 1144 (1955).

