The first review article in this series was published in the Bulletin on Narcotics last year and covered work which was published in the chemical literature through 1956 1.
Author: David Ginsburg
Pages: 1 to 5
Creation Date: 1958/01/01
The first review article in this series was published in the Bulletin on Narcotics last year and covered work which was published in the chemical literature through 1956 [1] .
The year 1957 has brought to the attention of chemists interested in the chemistry of the opium alkaloids a number of important papers. The three important contributions described below may be divided into several types: ( a) a paper which promises much in that it is the beginning of an investigation into the fate of morphine and codeine in the body; ( b) a series of papers which is the culmination of a very difficult structural elucidation; ( c) a continuation of the practical synthesis of potential analgesic compounds.
I. Degradative Studies directed towards the Synthesis of Labelled Morphine Derivatives
The fate of morphine and codeine in the human body has been the subject of much speculation, but to the knowledge of this reviewer no systematic organic chemical work has been undertaken heretofore in order to determine the identity of possible metabolic products of these alkaloids. It is there- fore good news that a master in the field of the opium alkaloids has undertaken preliminary research directed towards the study of morphine and codeine metabolism.
Rapoport, Chadha & Lovell [2] have studied the selective oxidation of the double bond in ring C of codeine and some of its derivatives. The aim was to cleave ring C and then to reconstitute it after replacement of one of the carbon atoms with radioactive carbon which might then permit tracing the metabolic fate of the reconstituted compound. At the same time, ring C glycols may be by analogy with cases known from other fields of organic chemistry and biochemistry (e.g., certain polycyclic hydrocarbons), metabolic intermediates in the biological degradation of codeine or morphine.
Codeine (I) was oxidized to the corresponding glycol (II) by means of osmium tetroxide at room temperature in ether solution and in the presence of pyridine. The resulting osmate was best cleaved by using ethanolic sodium sulfite, the over-all yield of the glycol being 60%.
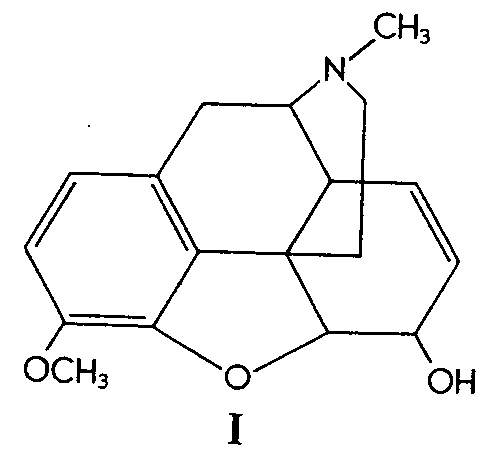
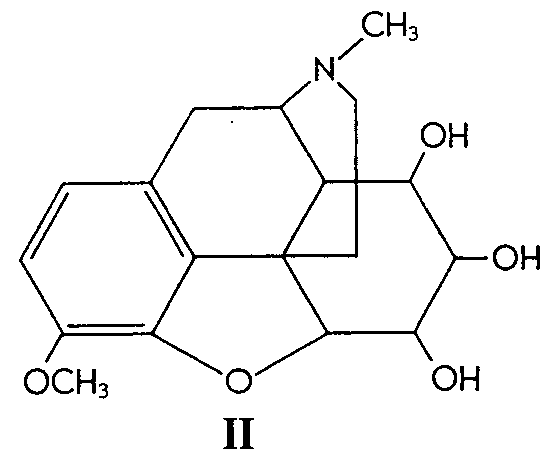
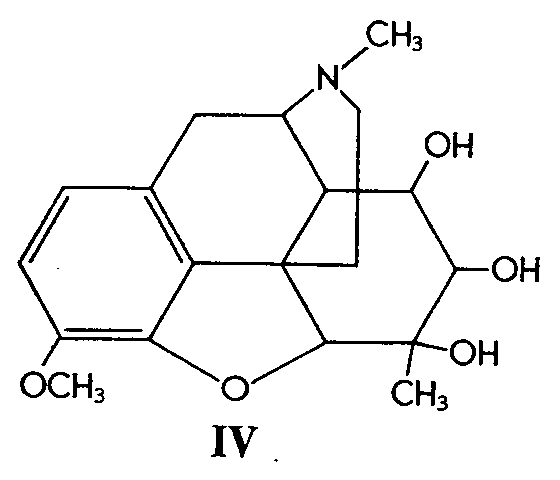
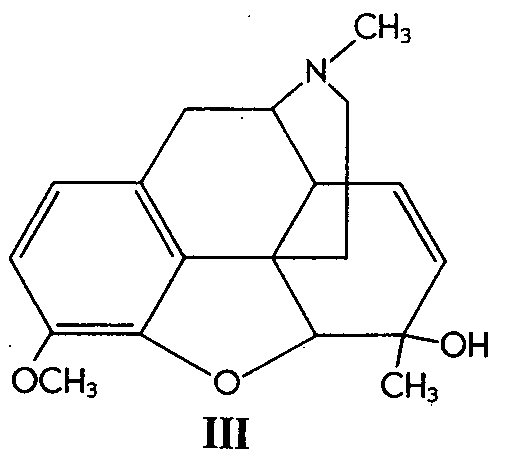
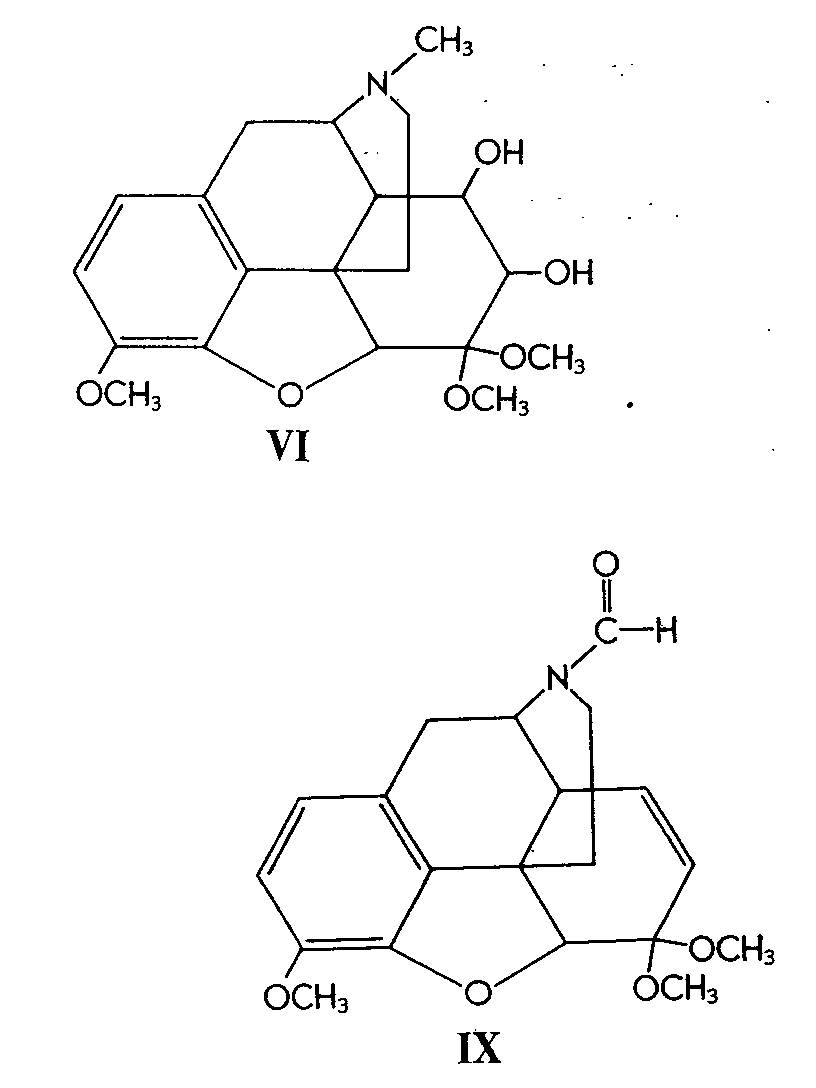
Similarly, 6-methylcodeine (III) gave the glycol (IV)in 52% yield. Codeinone dimethyl ketal (V) gave its corresponding glycol (VI) in 38% yield and &Delta[7] -desoxycodeine (VII) gave the glycol (VIII) in 92% yield.
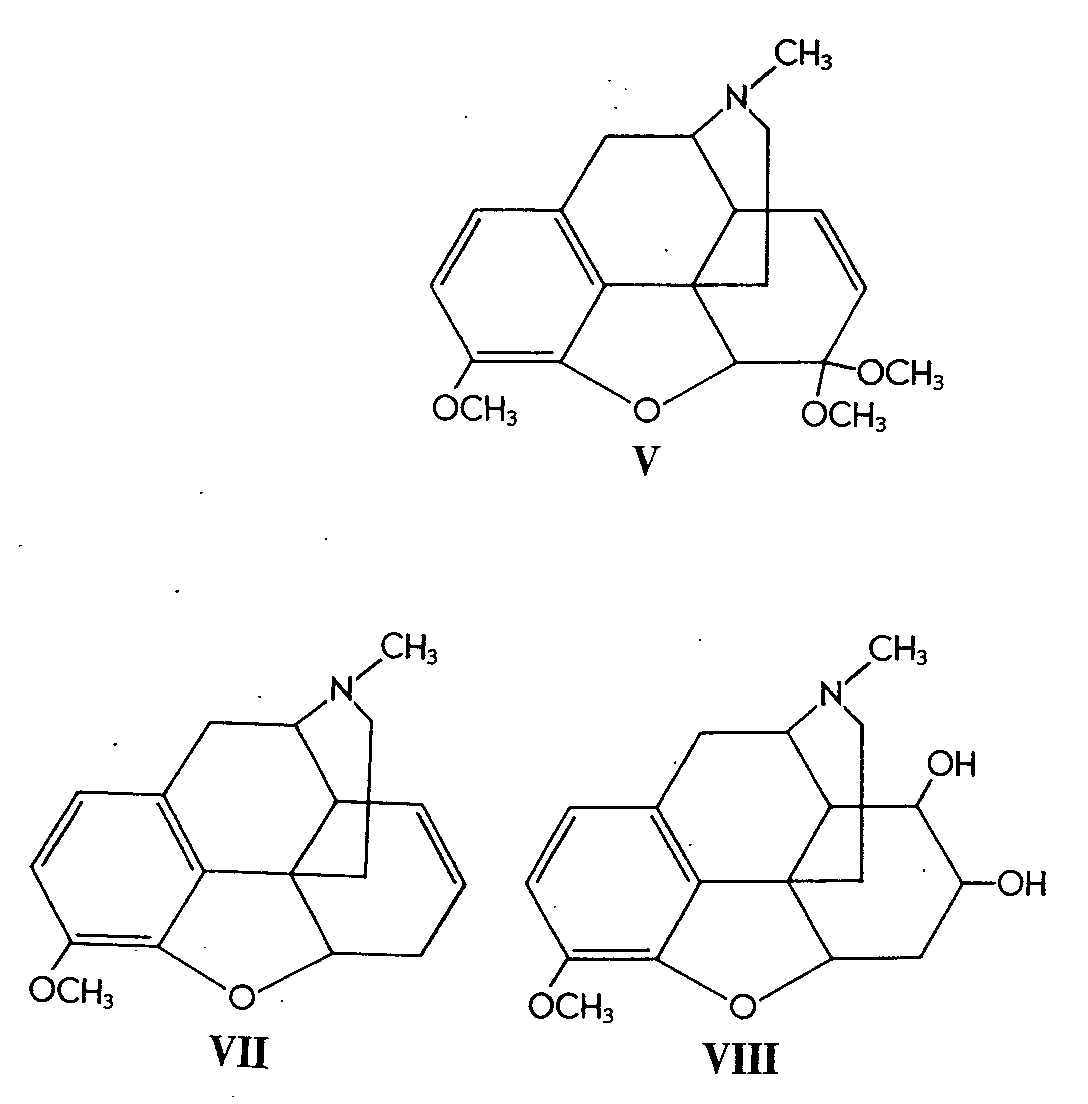
It is interesting to note that in the case of codeinone dimethyl ketal (V) a competitive reaction also took place -namely, oxidation by osmium tetroxide alpha to the nitrogen atom, affording N-formyl -norcodeinonedimethyl ketal (IX).
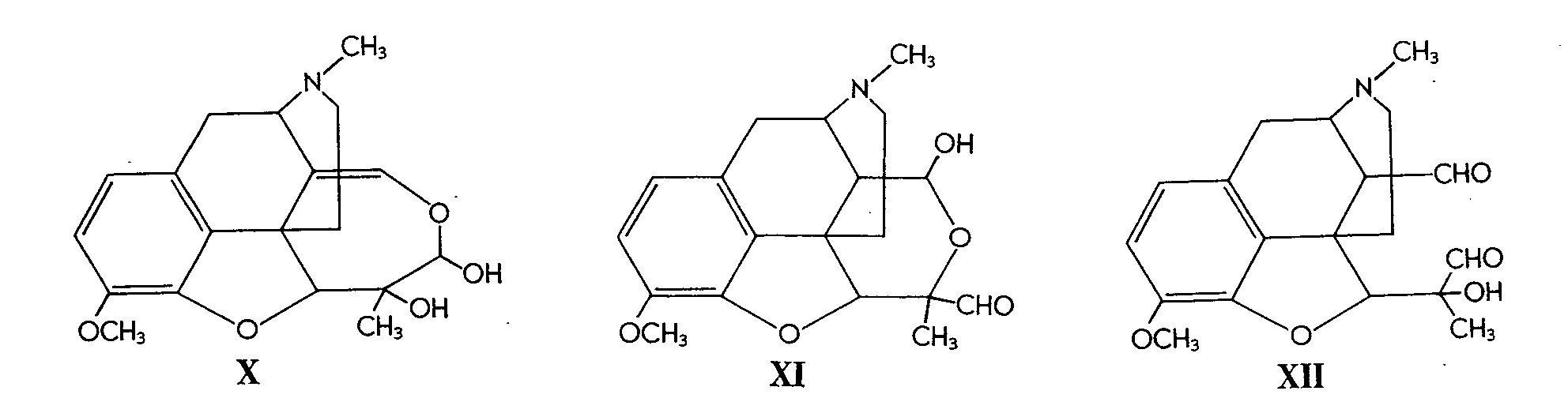
The glycols prepared were then further oxidized in order to cleave ring C. For example, upon oxidation in acid solution, IV consumed one mole of periodate, yielding various compounds formulated as X and XI (or XII).
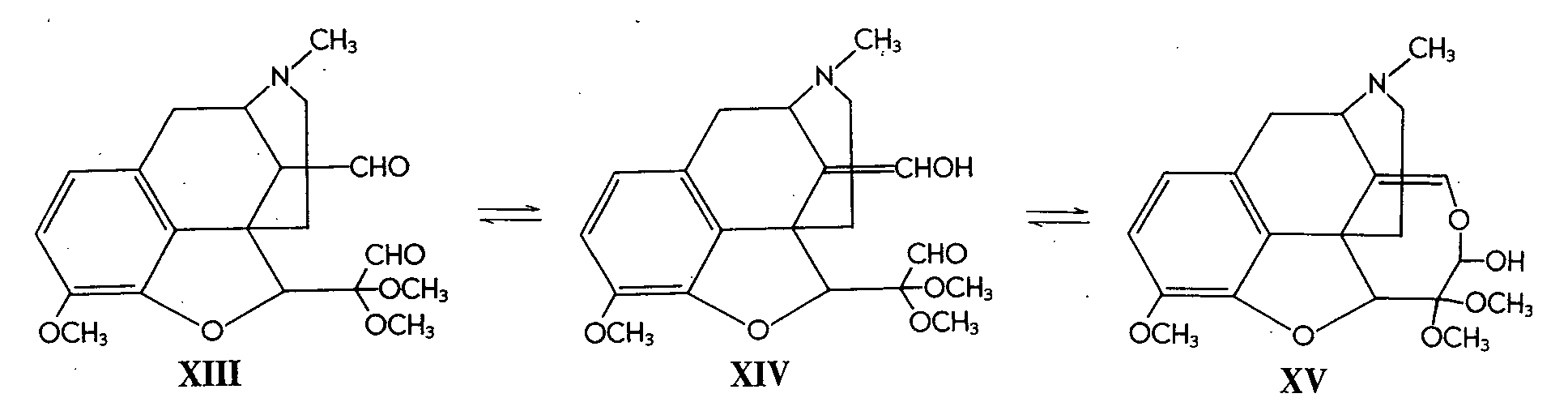
The hemiacetal formulations (X and XI) received further support from the products obtained by periodate oxidation of VI. Here again various products were obtained according to pH, and these were formulated as XIII, XIV and XV, which may be easily interconverted.
II . The Structure of Flavothebaone
Early in 1953 the reviewer was present at an Oxford Alembic Club lecture by Dr. J. Dominguez, who presented an account of work he had carried out on a reaction which at that time was very difficult to interpret. We now have before us an exhaustive research embodied in a series of four recent papers [3] which are the culmination of beautifully executed experimental work and brilliant deductive reasoning by Bentley and his co-workers. A very complex problem appears to have been solved, and convincing evidence and reasoning allow us to accept with confidence the following structure (XVII) for flavothebaone. The stereochemistry and mechanism for the rearrangement of thebainequinol (XVI) into flavothebaone are reasonably explained [4]
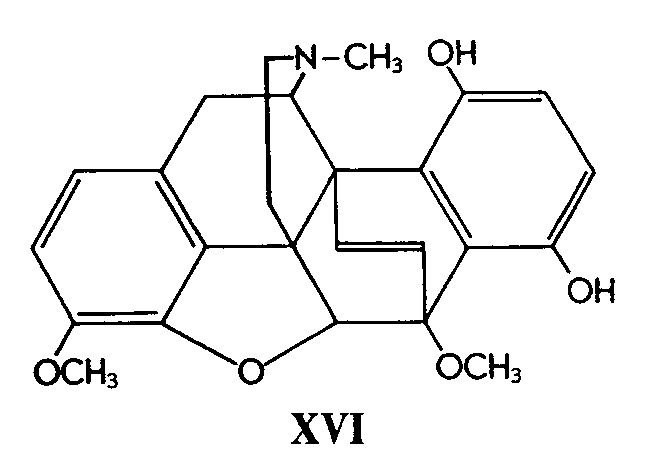
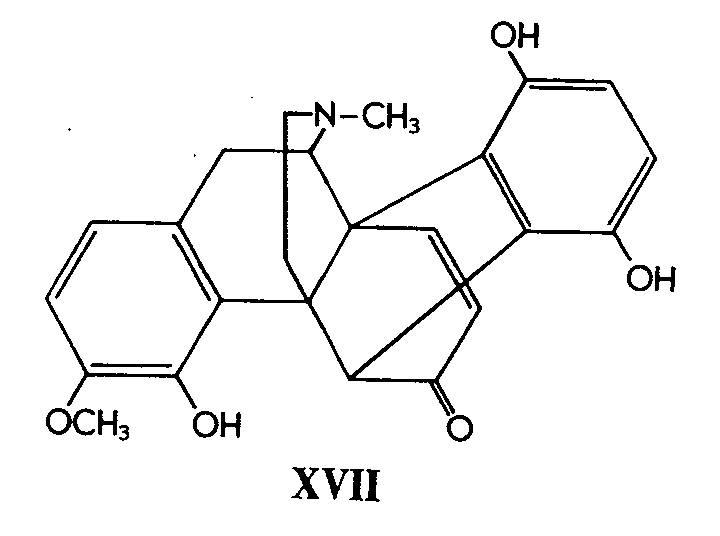
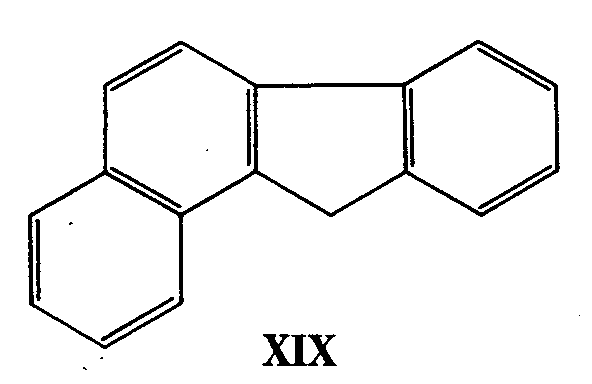
The complex series of transformations involved in the exhaustive methylation of flavothebaone trimethyl ether (XVIII) culminating with derivatives of the chrysofluorene ring system (XIX) has also been elucidated [3] .
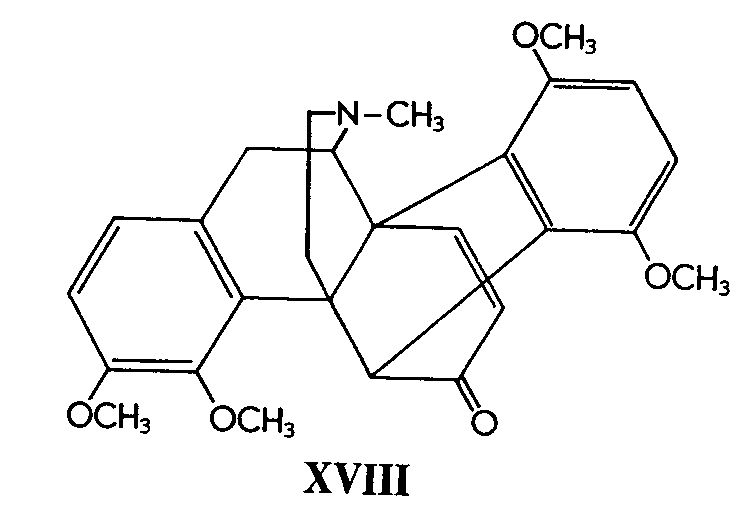
Fitting into the structural scheme in accordance with the above formulation for flavothebaone (XVII), various other flavothebaone derivatives have been degraded - e.g., to thebenone type compounds [5] and simpler derivatives, again believed to be based on the chrysofluorene skeleton [6] .
For the details of these investigations the reader is referred to the original papers. It is hoped that further interesting communications will soon be forthcoming from Dr. Bentley's Aberdeen laboratories, embodying the structural elucidation of other Diels-Alder adducts of thebaine.
III . Synthetic Morphinans
The Hoffman-La Roche laboratories at Basel have continued their synthesis of morphinan derivatives and have reported the preparation of several compounds characterized by extremely intense analgesic activity [7] .
Several synthetic approaches were used. The starting materials were either (-)- and (+)-l-( p-hyclroxybenzyl)-octahydroisoquinoline (XX) or (-)- and (+)-3-hydroxy-morphinan (XXIII).
1- p-hydroxybenzyl-1,2, 3, 4, 5, 6, 7, 8-octahydroisoquinoline (XX) was alkylated at the nitrogen atom and the N-alkyl derivative (XXI) was cyclized to the N-substituted morphinan (XXII).
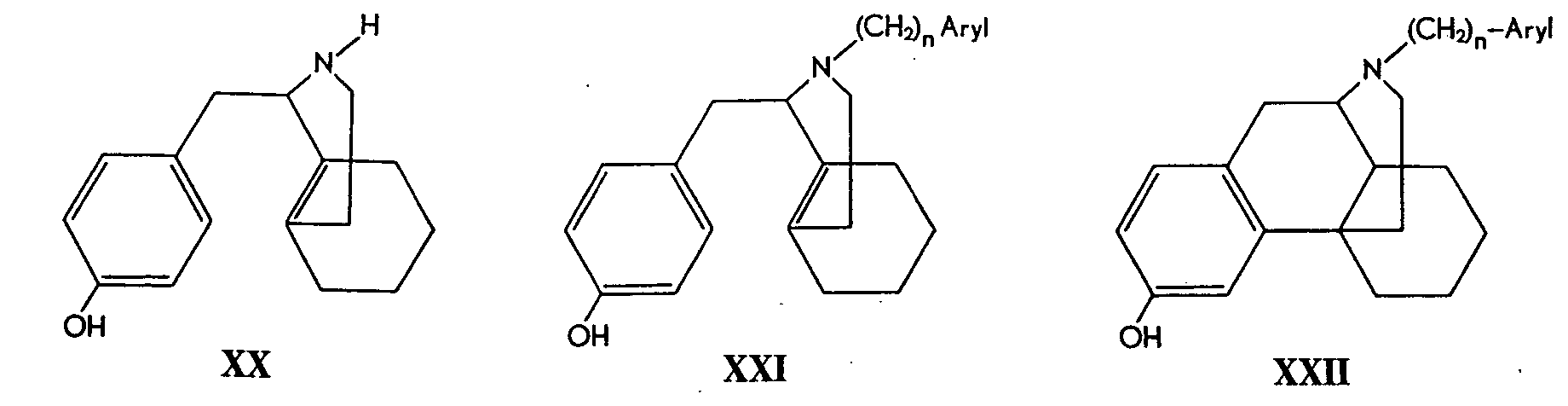
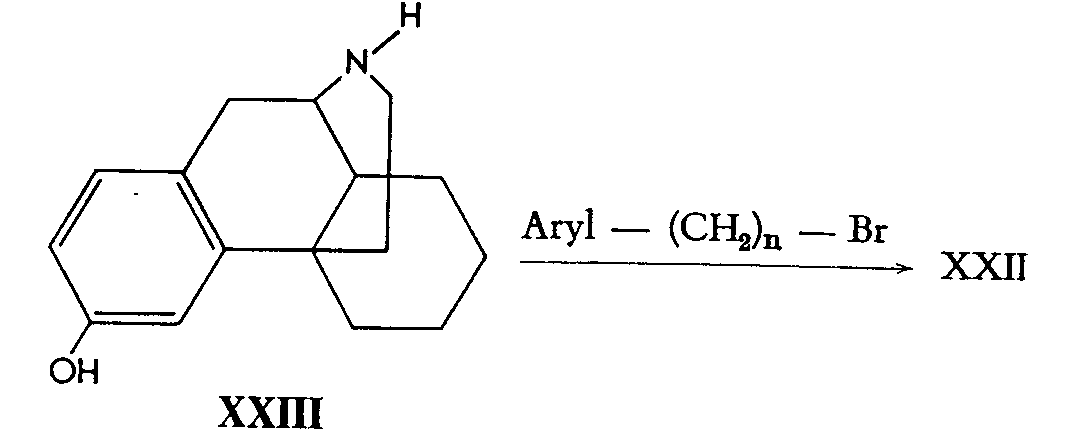
Alternatively, 3-hydroxymorphinan (XXIII) already containing the tetracyclic portion of the opium alkaloid skeleton was treated with arylalkyl bromides again yielding compounds of type XXII.

Another synthetic approach, using 3-hydroxymorphinan as the starting material, was treatment with arylalkyl acid chlorides. N-acyl-morphinans (XXIV) were thus obtained and these could be reduced to compounds of type XXII by reduction with lithium aluminium hydride.
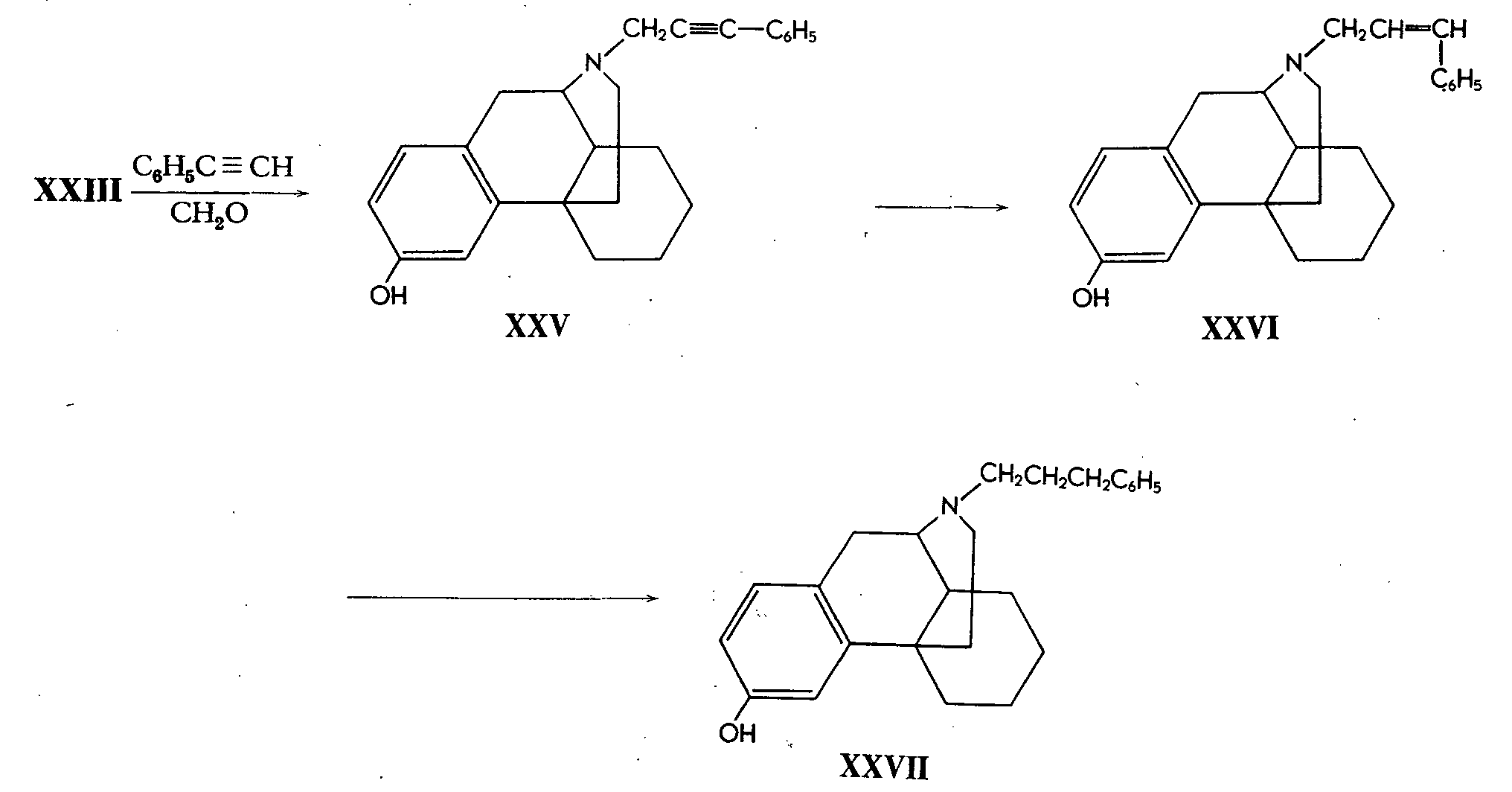
Other types of arylalkylmorphinans could be obtained by two additional routes. Treatment of (-)-3-hydroxy-morphinan with phenylaceteylene and formaldehyde affords (-)-3-hydroxy- N-phenylpropargyl-morphinan (XXV). Par- tial hydrogenation of the triple bond affords the N-phenyl-allyl derivative (XXVI) whilst its full hydrogenation again affords a compound of type XXII, namely the N-phenyl-propyl derivative (XXVII).
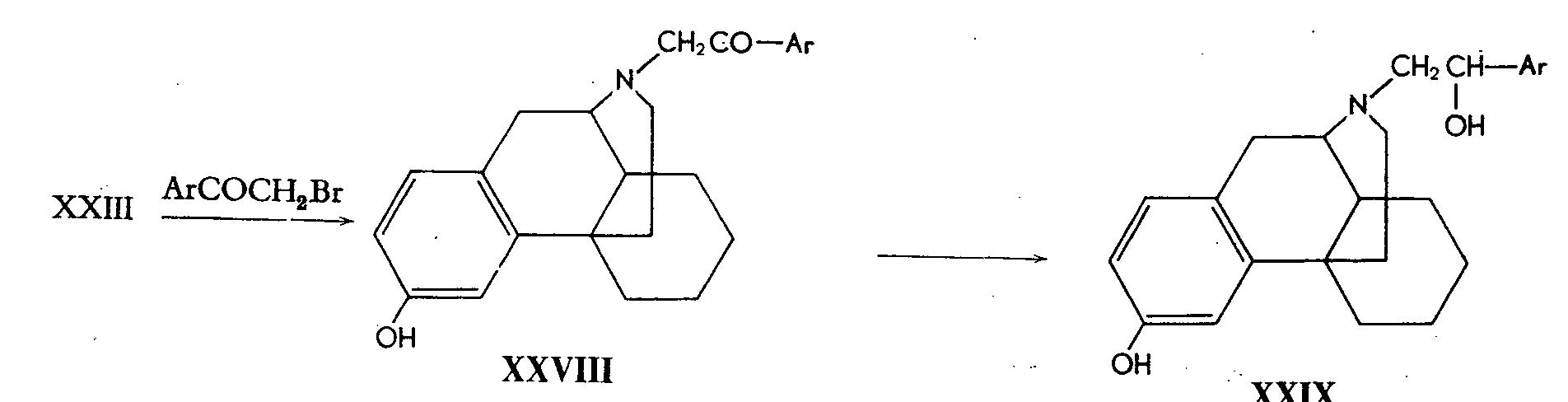

Treatment of 3-hydroxymorphinan with bromo-aceto-phenone or its homologs gives N- phenacyl derivatives (XXVIII) and these can be reduced to the N-acyl-hydroxy-alkyl compound (XXIX).
( - )-3-Hydroxy- N-phenylethyl-morphinan (XXX) and ( - )-3-hydroxy- N-phenacyl-morphinan (XXXI) and several of their derivatives showed 50-70 times the analgesic activity in animals as compared to morphine itself and are several times more effective than ( - )-3-hydroxy- N-methyl-morphinan (Dromoran).
References
001GINSBURG, Bulletin on Narcotics, Vol. IX. No. 3, 18 (1957).
002RAPOPORT, CHADHA & LOVELL, J. Amer. Chem. Soc., 79, 4694 (1957).
003BENTLEY, DOMINGUEZ & RINGE, J. Org. Chem., 22, 409 (1957).
004BENTLEY, DOMINGUEZ & RINGE, J. Org. Chem., 22, 418 (1957).
005BENTLEY, DOMINGUEZ & RINGE, J. Org. Chem., 22,422 (1957).
006BENTLEY & RINGE, J. Org. Chem., 22, 424 (1957).
007GR?SSNER, HELLERBACH & SCHNIDER, Helv. Chim. Acta, 40, 1232 (1957).

