Analysis of an impure heroin seizure
Introduction
Materials
Apparatus
Methods
Results
Discussion of results
Connexion of samples by chemical analysis
Summary
Acknowledgements
Author: T. W. McConnell Davis, Charles G. Farmilo, Klaus Genest
Pages: 47 to 57
Creation Date: 1962/01/01
A summary of this paper was presented to the 8th annual meeting of the Canadian Society of Forensic Sciences, Montreal University, Montreal, Quebec, 7 and 8 November 1960, by T. W. McConnell Davis.
Contribution from the Organic Chemistry and Narcotic Section, Food and Drug Directorate, Department of National Health and Welfare, Ottawa, Ontario, 8 November 1960.
Seven samples were submitted by the Royal Canadian Mounted Police Drug Squad to the analysts of the laboratories of the Food and Drug Directorate as exhibits in this narcotics seizure. The first sample (Lab. No. V-607) was purchased by the police undercover agent and brought to Ottawa. After identification of its narcotic and other constituents in V-607 had been established by physical and chemical methods of analysis, a certificate was issued. On the strength of this evidence, the police arranged to purchase a further 1? lb. through their agent. These samples were in turn submitted to us for assay.
Police circumstantial evidence showed that the heroin which was seized in this case was identical with that which is referred to as "crude granulated" heroin, and is the type used by the addicts "chasing the dragon" in Hong Kong [ 15] . Gruhzit [ 16] reported a heroin content of 80-99%, but gave no indication of the nature of the impurities. This is the first heroin seizure of this type to be smuggled into Canada from Hong Kong. Since it is of importance to narcotics authorities to try to establish by scientific methods origin connexions in cases of smuggled heroin, these results and discussion will be of international interest.
In this regard, it is of interest to quote an article [ 1] which appeared in the United Nations Bulletin on Narcotics: "The heroin traffic is an international one, but too little is known in any country about the characteristics of illicit heroin elsewhere. Knowledge of the adulterants found in illicit heroin in the various countries may be of value to enforcement officers everywhere. Accounts of other methods found of value for identification, determination, or particularly for detection of certain adulterants by direct tests on the heroin sample will be welcomed." In partial fulfillment of this request, it is the purpose of this paper to present the details of the reagents, apparatus, methods and results of the assay of the samples in this international narcotics case.
The samples consisted of pinkish-white granules about ? cm in diameter. The samples, along with their weights, are identified as follows: V-607 (43 g), V-684 A (253 g), V-684 B (205.8 g) V-684 C (202 g), V-686 (89.8 g) and V-687 (0.36 g). The latter sample was found on the person of the accused.
(A) Colour
Zernick's. Nitric acid (s.g. 1.42, 70%);
Marquis'. Dissolve 2 drops of formaldehyde (40%) in concentrated sulfuric acid (3 ml);
Frohde's. Dissolve sodium molybdate (0.5 g) in concentrated sulfuric acid (100 ml). The reagent is permanent when kept stoppered;
Mecke's. Dissolve selenious acid (0.5 g) in concentrated sulfuric acid (100 ml);
Ferric chloride. Dissolve ferric chloride (5 g)in water (l00 ml);
Kieffer's. Two solutions are prepared: (A) Dissolve potassium ferricyanide (0.05 g) in water (10 ml). (B) Dissolve ferric chloride (1 g) in water (10 ml). To solution A (10 ml) add 1 drop of solution B just before the reagent is required for use;
Oliver's. This is a test-tube reaction. Dissolve the unknown (1 mg) in water (1/2 ml) and add a trace of copper sulfate solution (1% in water) by means of a glass rod wetted with the copper sulfate. Stir. Add hydrogen peroxide (3%, 1 ml). Mix. Add concentrated ammonium hydroxide (1 ml). Shake. Observe the pink to red colour which persists for several hours with diamorphine. Morphine gives a transient red colour;
Murexide. Add a solution of the unknown (3 drops) to a micro crucible and dry carefully on a steam bath. Add bromine water (3-4 drops) evaporate on a steam bath. Cool. Expose the residue to ammonia fumes. The colour changes from orange to purple when caffeine or other xanthine alkaloids are present.
(B) Crystal
1:60 gold chloride and concentrated hydrochloric acid. This reagent is used for dry 0 6-monoacetylmorphine [ 1] . 1
The structural relationships of O6-and O3-monoacetylmorphines are shown in figure 9. 47
Rosettes of triangular plates, and single triangular plates are the characteristic forms;
Mercuric iodide in hydrochloric acid. Saturate dilute hydrochloric acid (1 plus 3) with mercuric iodide. This reagent is applied to a little of the dry substance containing heroin on a microscope slide. The heroin crystals are branching threads and splinter-plates [ 1] ;
Gold bromide in (2 plus 3) sulfuric acid. Dissolve gold chloride (HAuCl 4) crystals (1 g) in hydrobromic acid (40%, 1.5 ml) and (2 plus 3) sulfuric acid (24 ml) (a mixture previously made and cooled of 2 parts by volume of concentrated sulfuric acid and 3 parts by volume water). The crystals with heroin are fine needles, mostly scattered, but often partially in sheaves or rosettes. The interference of quinine is much diminished by the presence of acetic acid in the test solution. The test is very sensitive, and is best obtained on a very dilute solution, containing just enough diacetylmorphine for a light amorphous precipitate when the reagent is added. Caffeine yields somewhat similar crystals with this reagent;
Gold bromide in hydrobromic acid. Dissolve gold chloride (HAuCl 4) crystals (1 g) in (9 plus 1) hydrobromic acid (40%, 37.5 ml) with water. The reagent is applied to the dry powdered unknown suspected to contain caffeine. The caffeine forms needles immediately, and at the centre of greatest concentration red grains and prisms form gradually;
Silver nitrate. Silver nitrate (5 g) in water (100 ml). The test drop is acidulated with nitric acid. A white flocculent precipitate is indicative of the presence of halide ion.
(C) Chromatographic
Solvents. Iso-BuOH: AcOH: H 2O (10: 1: 2.4) is mixed and shaken until clear. This solution is used as the mobile phase. A small portion of the mobile phase is taken and shaken with water (100 ml) until it is water saturated. This aqueous layer is drawn off and used for equilibration of the chromatographic chamber.
Paper treatment. Whatman 3 MM paper (18" x 22") sheets are dipped in 0.5 M aqueous potassium dihydrogen phosphate (pH 4.2) and ammonium sulfate (2%, pH 5.3) dried and used for the preparation of the chromatograms;
Standard drug reference solutions. Reference standards of the narcotics and alkaloids are prepared by dissolving the base (50 mg) in methanol (10 ml) and stored at 4°C until required. The standards used for these assays were morphine, codeine, heroin and quinine in one solution and caffeine, 0 3-mono-,and O 6-monoacetylmorphine in a second solution. Lactose, procaine, thebaine and papaverine were combined in a third solution. Themalon ® was also included, since it is a thiambutene manufactured in Japan, and the seizure was suspected of originating in the Orient. The reference solutions were used as standards and spotted on the original exploratory chromatogram. (See spots Nos. 6, 7, 8, fig. 1.)
(D) Spray
Potassium iodoplatinate. Dissolve platinum chloride (10%, 1 ml) in potassium iodide (4%, 25 ml) and dilute with water to 50 ml. This is the general alkaloidal chromatographic spray reagent for opiates and synthetic narcotics which yield red to blue spots. Caffeine does not yield blue spots with this reagent;
Modified Dragendorff Ludy-Tenger (12). Bismuth subcarbonate (1 g) and potassium iodide (6 g) and hydrochloric acid (conc., 15 ml) were diluted with water to 100 ml. The reagent at this concentration may be further diluted (1 : 1) with water, if desired for respraying faded chromatograms. Caffeine and other alkaloids yield orange spots.
Kieffer. The potassium ferricyanide reagent described in section A (f) above was used for spraying the chromatograms to detect the O 6-monoacetylmorphine. Blue spots are produced with monoacetylmorphine.
(A) Glassware
Spot plates, micro test tubes, tapered glass rods, microscope slides, microcrucibles, microspatulas;
Semi-micro Soxhlet extractor provided with the Quickfit ® semi-micro-organic preparation apparatus. This set is equipped with filtration funnels and other devices convenient for handling milligramme amounts of materials;
Chromatographic jars (9 gal. capacity, 24" x 12"). Spray reagent bottles, spotting pipettes, lambda type, 5, 10, 20 µl capacity.
(B) Instruments
Ultraviolet lights, 2537 and 3660 ?;
Polarizing microscope and accessories;
Ratio recording spectrophotometer equipped with 1 ml capacity, 10 mm pathlength quartz cells;
pH meter;
IR. spectrophotometer, equipped with sodium chloride cavity cells, 0.5 mm pathlength.
The seven samples were given a careful physical examination. The weights were recorded, and a portion of each was ground to a fine powder. A small amount (1 mg) was placed in a micro test tube, dissolved in water (3 drops), and about three drops of phosphomolybdic acid reagent were added. In each case, a heavy flocculent precipitate was observed. Small amounts of the powder from each sample were placed in the depressions of several porcelain spot-test plates. The colour test reagents: Zernick's, Marquis', Frohde's, Mecke's, ferric chloride, Kieffer's, were added consecutively to the unknown, and to samples of heroin, and O 6-monoacetylmorphine as standards for comparison of the colours produced. The colour changes were observed at timed intervals. Microcrystal tests were carried out on each of the samples either on the dry powder, or on very dilute aqueous solutions of the unknowns, depending on the type of test. The crystals were observed using a polariz-ing microscope. A record of the crystal forms was made. Immediately on receiving the samples, preparation of solutions for chromatography was begun. This is a routine procedure for the investigation of all narcotic cases received in this laboratory.

Chromatography. - The sample (100 mg) was dissolved in a few drops of methanol in a 10 ml volumetric flask, and 10 ml chloroform was added to make to volume. The chromatogram was prepared as illustrated in figure 1. The first four spots are heroin standards at 100, 80, 60 and 40 ?g respectively. On the fifth spot was placed 400 ?g (approx.) of a methanol mother liquor from sample V-607. Spots 6, 7 and 8 contained the standards combined so that the spots did not interfere with each other. Spot 9 contained the sample V-607, and spots 10 and 11 were locations of the two residues from the mother liquor prepared by recrystallization of the sample V-607. Spot 12 is the location of the synthetic narcotic, Themalon ®.
Chromatography was carried out in accordance with the procedures outlined in previous articles [ 2] , [ 3] , except that the equilibration step is omitted to shorten the time of analysis. This required the use of standard reference compounds for comparison of Rf-values. It is necessary to include the equilibration step for reproducible Rf-values. The total time for the determination was 8 hours. 2 The analyst's time for preparation and spraying and examination was ? hour.
A number of chromatograms were prepared as shown in figure 2 A for separation of larger amounts of the components of the seven samples for elution and assay. A 34-cm line was left, along which the solution of the unknown containing 5 mg of material was streaked evenly along the starting line of the chromatogram. The equilibration and development procedures were carried out [ 2] , [ 3] .
After drying to remove the solvent, the chromatogram was examined under ultraviolet light, the solvent front marked and the UV absorbing areas carefully outlined to include all irregularities.
Chromatograms were prepared for spraying according to the procedure illustrated in figure 2. The exposed areas in figure 2 A were then sprayed with the potassium iodoplatinate, except for the caffeine spot, which had been covered. The caffeine areas were then uncovered (see figure 2 B). The chromatogram was sprayed. This time the modified Dragen-dorff reagent was used (12). The caffeine spots produced a bright orange colour. The chromatogram was then examined carefully, and the boundaries of the stained areas of the three components were marked. Straight lines were then drawn horizontally across the chromatogram. There were three areas of paper which were then cut out and sliced into fine shreds, transferred to separate Erlenmeyer flasks and eluted.
________
A method (system 3) requiring a total of 5? hours elapsed time (l? man-hours technicians' time) for 14-17 samples has been described by Genest & Farmilo [ 18] .

The chromatogram in each Erlenmeyer was eluted with 50 ml portions of the series of solvents given in table 1.
For each elution step, 50 ml of the solvent were added, and the paper and solvent warmed on the water bath with occasional shaking for ? hr. At this time, the solvent was poured into a weighed evaporating dish, and a portion of the new solvent was added and the procedure repeated. In this manner, the elution with each of the five eluting solvents was carried out. Finally, the solvents were evaporated and the residues were weighed. A blank from a region of the chromatogram free of alkaloids was made in the same way. The residues were then assayed by means of ultraviolet spectrophotometry.
|
No. |
Volume |
Solvent |
Ratio of components |
|---|---|---|---|
| 1 | 50 |
Chloroform: Ethanol
|
2:1
|
| 2 | 50 |
Chloroform: Ehanol
|
1:2
|
| 3 | 50 |
Ethanol
|
|
| 4 | 50 |
Ethanol: Water
|
1:1
|
| 5 | 50 |
Water
|
|
Compound |
Max.m?. |
Emax. |
Solvent |
pH |
Ret. |
|---|---|---|---|---|---|
|
Caffeine
|
273 | 7,770 | 4 | ||
| 278 | 10,300 |
Water
|
7 | 5 | |
|
Heroin
|
281 | 1,940 |
Ethanol 95%
|
-
|
6 |
|
O6-monoacetylmorphine.
|
287 | 1,460 |
Ethanol 95%-
|
-
|
6 |
For the purpose of assay of the eluates, the concentration-absorbances relationships were determined for each of the substances, using the data given in table 2 and substituting in the general equation:
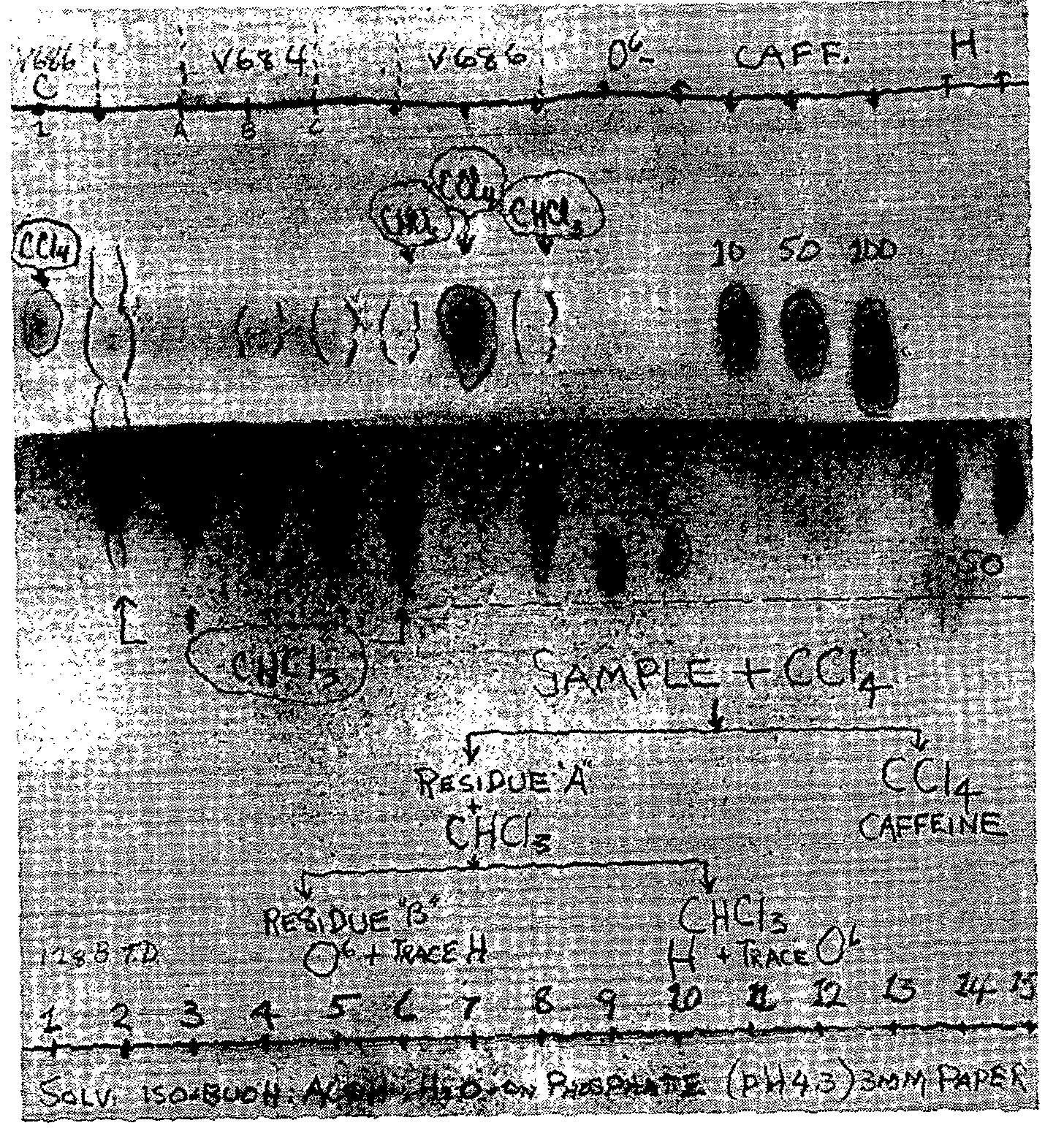
(1) concentration,
C (grammes per litre) = absorbance x molecular weigt / molecular extinction value, which procedure yields the specific relationships:
(2) C = 0.025 ? A273m ?for caffeine
(3) C = 0.19 ? A281m ?for heroin, and
(4) C = 0.224 ? A287m ?for O 6-monoacetylmorphine.
Example of calculation
For the sample V-684C, the absorbances for O 6-monoacetylmorphine at 287 and 310 m? were subtracted: (5)... A 287-A 310= 0.14. Substituting this value in equation (4) above, C = 0.224 x 0.14 = 0.0314 g/l, which corresponds to 0.157 mg in 5 ml. Since a portion of the O 6-monoacetylmorphine is used in spray-detection of the outline of the band, it is necessary to make a correction for the loss of this material. The total length of the area of chromatogram eluted was 36 cm, of which 32 cm was taken for elution. An approximate correction factor is therefore 36/32, assuming a uniform distribution of substance along this strip. The corrected concentration is 0.157 x 36/32 = 0.176. The total weight of sample applied was 5 mg. The percentage amount of O 6-monoacetylmorphine present was 0.176/5 x 100 = 3.52%. Similar calculations were made for caffeine, and heroin.
The sample (1 g) was placed in an extraction thimble (35 x 10 mm) in a semi-micro Soxhlet extraction apparatus, and the boiler flask filled with carbon tetrachloride. The sample was extracted for ? hr. The solvent was poured into a clean, dry dish, evaporated and weighed. Fresh solvent was added to the boiler flask and the extraction process continued. Caffeine was observed to form a white, needle-filled rime above the surface of the liquid in the flask, and this rime diminished as the endpoint of the caffeine extraction was reached. The glassware was washed with a small amount of chloroform after the carbon tetrachloride had been poured into the weighing dish. The solvent was evaporated and the residue weighed after drying at 105°C. The weight was taken as the amount of caffeine present. The remaining powder in the thimble was then extracted with chloroform. The solvent was collected in another weighing dish, and after three extractions the combined solvent was evaporated, and the residue dried as above and weighed. This weight was taken as the amount of heroin hydrochloride present in the sample. The residue in the thimble was dried at 105°C and weighed. This weight was taken as the amount of O 6-monoacetylmorphine hydrochloride present in the original sample.
|
Conditions |
Caffeine |
Heroin HCl |
O6-monoacetylmorphine |
|---|---|---|---|
|
Origin (1)
|
From MEOH mother liquor of V-607
|
Residue from V-686-3H, CHCl
3 ext. oven-dried 105°C
|
Free base supplied by Dr. L. Welsh, F.D.A., Washington, U.S.A.
|
| (2) |
Caffeine alkaloid from E. Merck, Darmstadt, Germany
|
T. & H. Smith, Edinburgh, Scot- land
|
V-607, residue from 1 gm. sample
|
|
Phase (1)
|
Chloroform
|
Chloroform
|
Chloroform
|
| (2) | |||
|
Thickness (1)
|
0.5 mm cavity cells
|
0.5 mm cavity cells
|
0.5 mm cavity cells
|
| (2) |
"
|
"
|
"
|
|
Concentration (1)
|
0.0285/5 ml
|
. . .
|
5 mg/0.25 ml
|
| (2) |
0.050/5 ml
|
0.05/5 ml
|
. . .
|
|
Prism (1)
|
NaCl
|
NaCl
|
NaCl
|
| (2) |

The O 6-monoacetylmorphine-free base was isolated from the hydrochloride as follows: The salt in the thimble was washed with water into a separatory funnel. A few drops of concentrated ammonium hydroxide were added to the aqueous layer and the liquid extracted with chloroform immediately. The chloroform layer was drawn off through a pledget of cotton wool in the stem of the separatory funnel. The chloroform was caught in a weighed dish, the chloroform was evaporated and the residue weighed. This weight was taken as the amount of O 6-monoacetylmorphine-free base present in the sample. The sample was re-dissolved in the appropriate amount of chloroform and the solution used for infrared spectrophotometric measurement.
Identification of Constituents by IR
The infrared spectra of the compounds were obtained in chloroform solution using an automatic recording spectro-photometer. Pertinent details of the method are given in table 3.
The results of the colour tests with the samples V-607, V-684A, V-684B, V-684C, V-687, heroin hydrochloride,
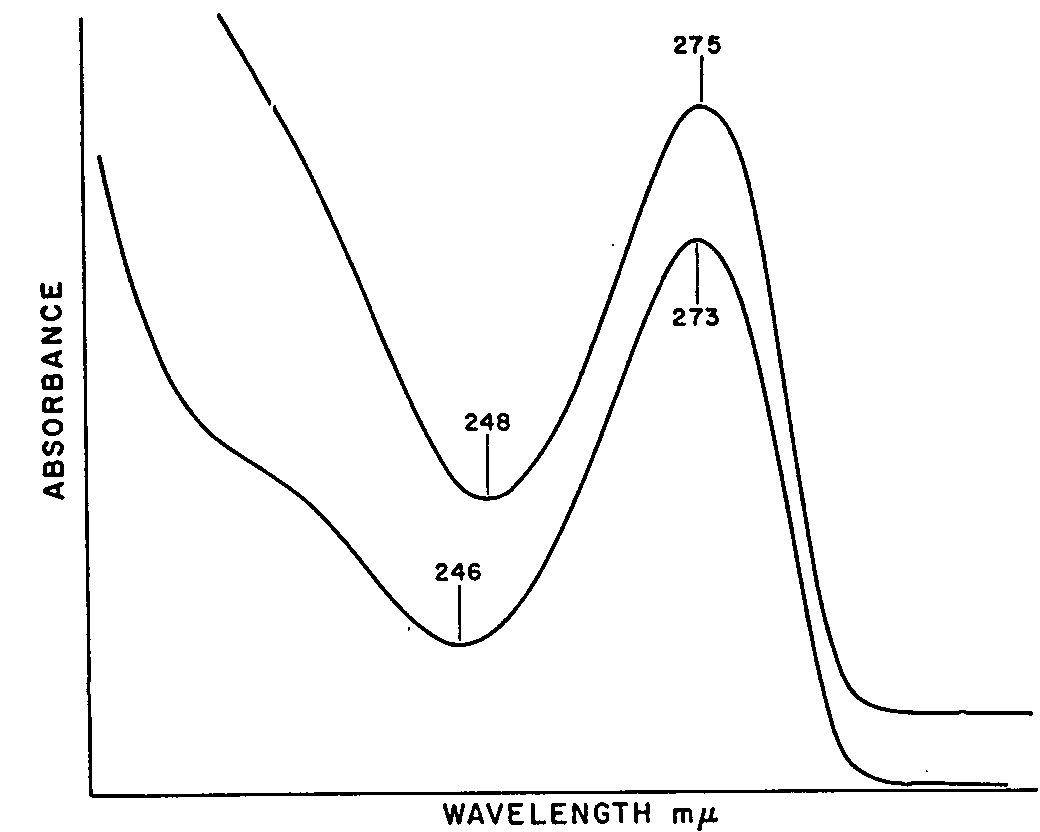
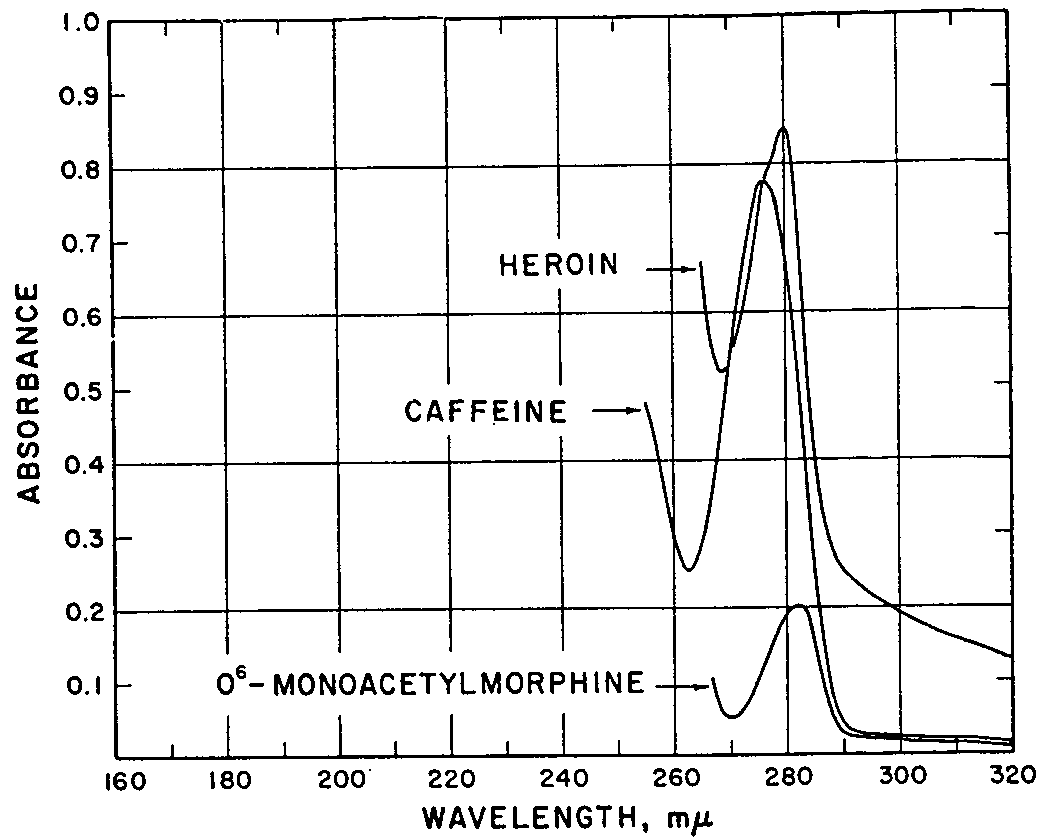
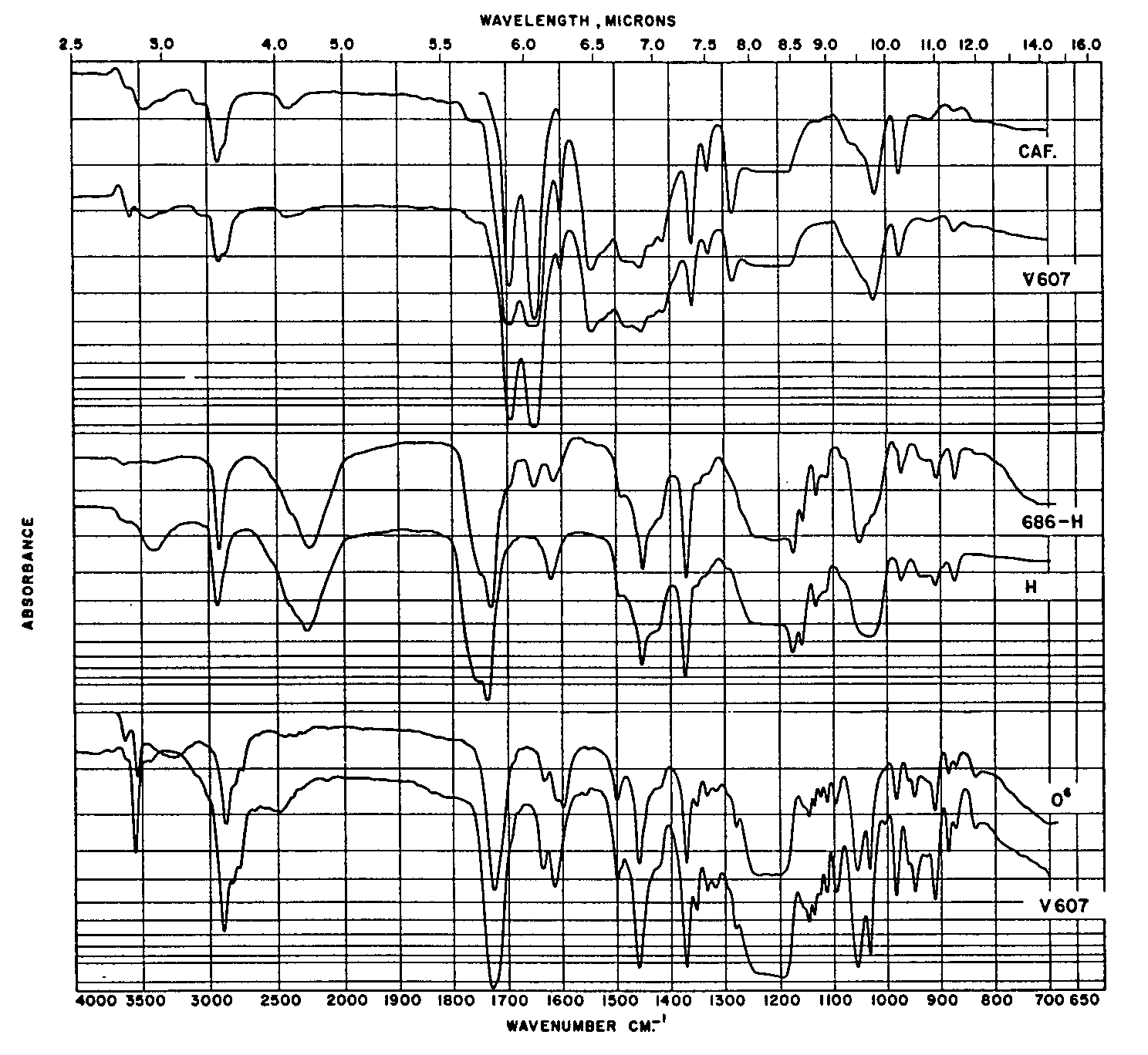
O 6-monoacetylmorphine ,and caffeine are given in table 4. In column 1 are listed the colour reagents by name, and in columns 2-4 inclusive are coded the colour changes that are observed with the lapse of time. In the fifth column are listed the time intervals in minutes. The last column lists the remarks and colour code. The results of the chromatographic experiments are shown in figures 1 to 5. Figure 1 shows the distribution of Rf-values obtained when the unknowns are chromatographed and compared with the standards. Spot 9 carried the unknown V-607. Figure 2 illustrates the arrangement of the chromatographic sheet for elution study, and the quantitative determination of the caffeine, heroin, and O 6-monoacetylmorphine by UV-spectrophotometry of the eluates. Figure 3 illustrates the combined assay sheet for the semi-quantitative estimation of the amounts of the constituents of sample V-607. The standards are applied in graded amounts, caffeine from 2.5 to 150?g, heroin from 20 to 120 ?g, and O 6-monoacetylmorphine from 5 to 10?g. The unknown V-607 is applied at the 200 ?g level. After staining, the density of the colours is compared visually, and the best-matching density is obtained. Figure 4 illustrates both the flow-sheet of the extraction method, and the efficiency of this method tested by chromatographic separation of the various residues. Reference standards of O 6-monoacetylmorphine, caffeine and heroin hydrochloride were chromatographed along with the residues from samples V-684-A, B and C, and V-686. Figure 5 illustrates a further chromatographic study of efficiency of the solvent extraction separation of O 6-monoacetylmorphine from caffeine and heroin. In addition, it shows the procedure for preparative chromatography in which milligramme quantities of the unknown V-607 are separated into the constituents, and the edge-spraying technique for chromogenic detection of the unknowns followed by cutting and marking of the areas for elution and further study by other methods. Figures 6 and 7show the ultraviolet spectra of the original sample from 340 to 220 m? dissolved in water, and of eluates from a preparative chromatogram such as the one illustrated in figure 5. Figure 7 is the spectrum of three separated substances taken from 360-230 m?. Figure 8 shows the infrared spectra of standards and isolates from various samples. They are paired to facilitate visual comparison of the maxima. The illustration is divided into three sections. In section one are the caffeine reference spectrum and that of the crystals from the methanol mother liquor from V-607. In the second section are the spectra of the isolate of heroin from sample V-686 made by the solvent extraction procedure, and a standard of heroin hydrochloride. In section 3, the spectrum of the residue which remained after extraction with carbon tetrachloride and chloroform is compared as the free base with the spectrum of O6 - monoacetylmorphine. Table 5 lists the major band types and their fre- quencies from the IR spectra of the standards and materials isolated from the samples. Table 6 lists the quantitative results obtained by a combination of the extraction and chromatographic elution procedures combined with UV-spectrophotometric assay of the eluates. Averages of values obtained for water and the minor constituent O 6-monoacetylmorphine are given, not the individual values for each sample.
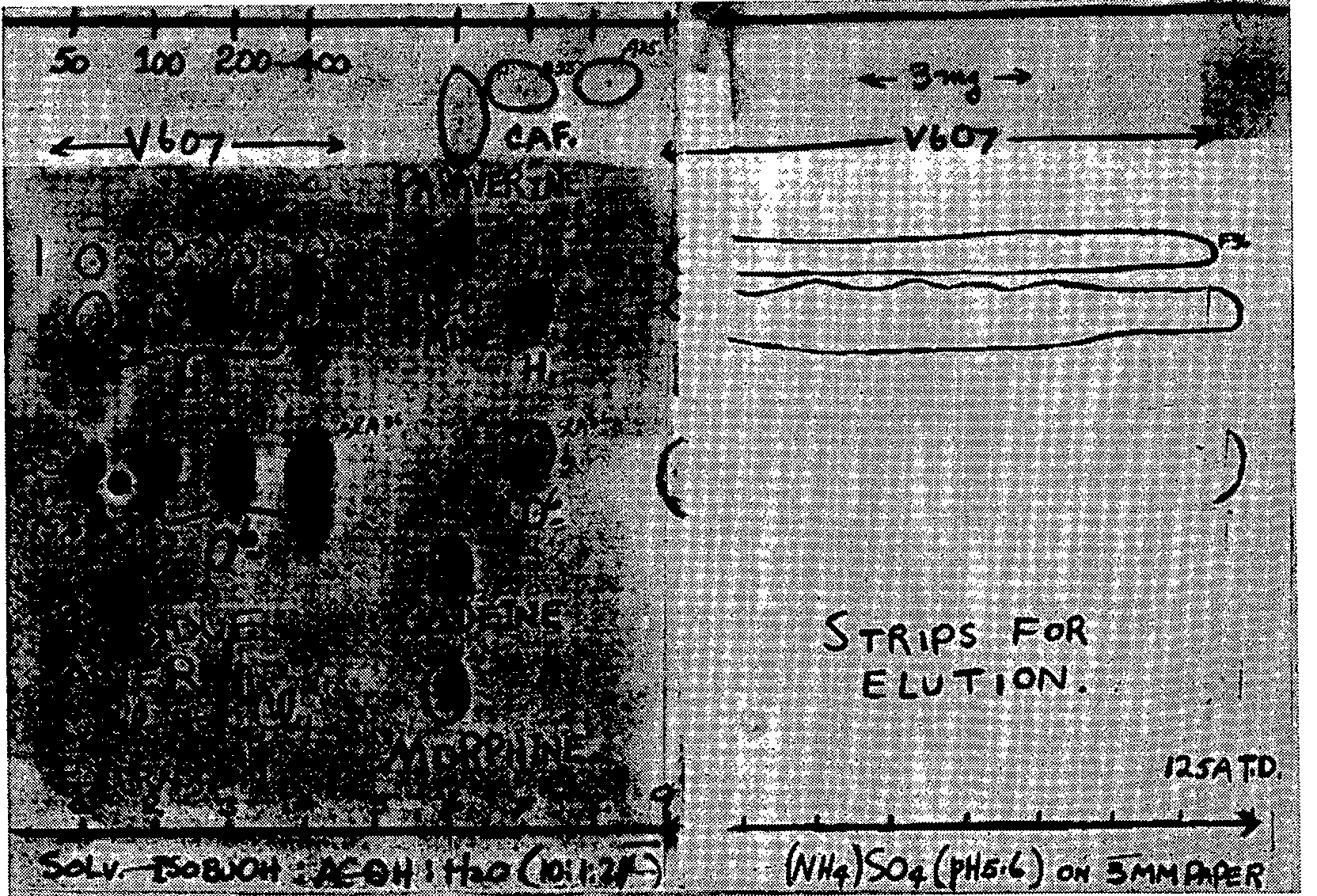
|
Colour observed with samples and standards |
|||||
|---|---|---|---|---|---|
|
Name of test reagent |
Un-knowns |
H a |
O 6 b |
Time (mins.) |
Remarks and colour code |
|
Zernick
|
Y
|
Y
L
|
O
VD
|
0 |
Primaries:
|
|
YD
|
Y
|
O
L
|
1 |
R = Red
|
|
|
G
|
G
c
|
O
Y
|
6 |
O = Orange
|
|
|
Y
|
Y
d
|
YD
|
20 |
Y = Yellow
|
|
|
Marquis
|
P
D
|
P
D
|
Nil
|
0 |
G = Green
|
|
P
|
P
|
P
|
1 |
B= Blue
|
|
|
B
P
|
B
P
|
B
P
|
3 |
V = Violet
|
|
|
B
|
B
|
B
|
6 |
P = Purple
|
|
|
Frohde
|
BV
|
V
|
V
L
|
0 | |
|
Gy
|
-
|
-
|
1 |
Tints and shades by Super Script:
|
|
|
G
|
G
|
Gy
|
2 | ||
|
B
c
|
B
|
B
c
|
4 |
L = Light
|
|
|
Mecke
|
G
|
G
|
B
|
0 |
D = Dark
|
|
BG
|
BG
|
G
|
0.5 |
VL = Very light
|
|
|
G
D
|
G
|
-
|
1 |
VD = Very Dark
|
|
|
G
VD
|
G
VD
|
G
VD
|
10 | ||
|
Mixed colours:
|
|||||
|
Ferricchloride
|
NC
|
NC
|
YD
|
0 | |
|
Y
|
Y
|
B
|
1 |
BG = Blue Green
|
|
|
Y
|
Y
|
BG
|
10 |
(blue is irst and dominant tone)
|
|
|
Y
|
Y
|
R
c
|
20 | ||
|
Kieffer
|
G
|
G
|
G
|
0 |
Gy = Grey
|
|
-
|
G
L
|
-
|
10 |
NC = No colour
|
|
|
Oliver
|
R
|
R
|
R
|
0 |
Caffeine: Y?O?P
|
|
R
|
R
|
R
|
10 | ||
|
Murexide
|
Y
|
Y
|
Y
|
0 | |
|
P,O
|
O
|
O
|
5 | ||
H = heroin,
bO6= O6-monoacetylmorphine.
cRim.
dFades.
|
Band type and frequency, cm-I |
||||
|---|---|---|---|---|
|
Substance Group |
OH |
N |
HC1 |
C=0 |
|
Heroin HCl
|
-
|
2,930 | 2,265 |
1,745-55
|
| 1,732 | ||||
|
O6-monoacetylmorpine.
|
3,540 | 2,885 |
-
|
1,728 |
|
Caffeine
|
-
|
2,930 |
-
|
1,695 |
| 1,650 | ||||
|
Sample number |
||||||
|---|---|---|---|---|---|---|
|
Substance |
V-607 |
V-684 A |
V-684 B |
V-684 C |
V-686 |
V-687 |
|
Heroin HCl and O6-monoacetyl-morphine
a
|
68.5 | 68.8 | 68.8 | 68.5 | 68.7 | (70) |
|
Caffeine and water
b
|
30.0 | 30.2 | 30.9 | 30.0 | 30.5 | (30) |
Average value, 3.6%
bAverage value, 3.8%.
Presumptive Identity of Sample Components
In the examination of the sample carried out by J. Ouellet, Food and Drug Directorate of Montreal, it was immediately observed that the colour tests and the ultraviolet absorption spectrum were unusual when compared with pure heroin or morphine. The Marquis test gave an unusual initial colour, more red than purple. The nitric acid reaction 3 gave a yellow colour which failed to change to green. It was later observed
Gruhzit [ 16] showed that the nitric acid test on presumably similar material required heating to produce a light green colour. U.S. narcotic chemists [ 17] report that the depth of green colour is an indication of purity, but make no mention of heating.
by the authors that with large quantities (more than 2 mg) the nitric acid reagent produced a faint green colour which faded to a yellow on standing for a short time. This unusual reactivity toward colour reagents and the somewhat unfamiliar shape of the UV-spectrum, figure 6, led Ouellet to conclude that an opiate was probably present. However, further work was required.
The red colour produced by Marquis' reagent was somewhat reminiscent of the shade of colour produced by some phenothiazines and antihistamines. These drugs have been reported to be used by addicts suffering a shortage of heroin [ 1] , [ 2] and [ 19] . The presence of a phenothiazine was excluded by a negative test for sulfur using the iodine-azide reaction described by Feigl [ 7] . Fluorescence tests for quinine in both neutral and acid (H 2SO 4)solution were negative. Fehling's test for lactose was also negative. However, positive silver nitrate and phos-phomolybdic acid tests were obtained. These tests indicated the presence of a hydrochloride of a nitrogenous base.
It can be seen in figure 1 that the unknown (spot 9) contains three components, which are by comparison with the standards (spots 1-4, and spot 7) tentatively identified as O 6-monoacetyl-morphine, heroin, and a third compound. The first two compare closely in colour and position (Rf-values of 0.42 and 0.58 respectively) with the standards. There are some other standards - e.g., procaine, O 3-monoacetylmorphine, quinine -on this paper which are similar to the unknown components, which were eliminated by further testing. The third compound, Rf-value 0.91, gave no colour with iodoplatinate (unlike any of the standards) and showed a strong UV-quenching effect with the 2537 ? emission when examined by transmitted light. None of the standards on the paper gave this set of properties. A comparison with tabulated chromatographic properties (Rf-values, chromogenic and UV-behaviour) revealed a close similarity between the unknown and caffeine. A murexide test on the original powdered sample (see table 4) confirmed their identity.
The presumptive identification of O 6-monoacetylmorphine, heroin hydrochloride and caffeine at this stage led to the necessity to develop an isolation procedure which would separate the weak base caffeine from the opiates. In the detailed analysis of heroin given by Fulton [ 1] , a method based on chloroform extraction from hydrochloric acid solution devised chiefly as a means of separating heroin from quinine was described. It is one of the features of this method that the heroin hydrochloride is extracted from the hydrochlorides of other alkaloids by its preferential solubility in chloroform. It was not desirable to use this method directly, since O 6-monoacetylmorphine hydrochloride and the heroin hydrochloride could hydrolize to morphine, as shown by paper chromatographic study of this method [ 1] . Furthermore, the excess of caffeine complicated the extraction procedure because of its solubility in chloroform. It was known that heroin hydrochloride and caffeine could be separated from the monoacetylmorphine after a preliminary experiment in which the powdered sample V-607 was washed on a filter with hot chloroform, leaving a Marquis-positive substance. Chromatography of this residue showed it to be mainly monoacetylmorphine. Further study of other chlorinated hydrocarbon solvents, trichlorethylene and carbon tetrachloride along with benzene and ether, was carried out. It was found that carbon tetrachloride dissolved caffeine, leaving the hydrochlorides of heroin and monoacetylmorphine. Combining the steps (i.e., extraction with carbon tetrachloride followed by chloroform) led to a method of separation of the three components. The efficiency of the extraction was then studied by means of paper chromatography of the extracts and residues. These results show a fairly efficient separation. Infrared spectrophotometry of the residues indicate less than 5% impurities.
Wright [ 8] and Welsh [ 9] have both written of the difficulty of preparing a crystalline hydrochloride of O 6-monoacetylmorphine. It is interesting to note that the non-aqueous extraction procedure outlined in this paper provides such a method. It should also be noted that the hydrochloride derivative cannot, because of its chloroform insolubility, be studied in this solvent by IR-spectrophotometry. In some experiments, the residue was left as a sticky resin which gradually hardened on standing in a desiccator.
It is well known that in the Stas-Otto procedure the chloroform residue may contain bases which have been extracted as the salt from acidic solution. Umberger [ 10] points out that a solution to the problem of base carry - over may lie in the use of carbon tetrachloride as an extractant at this stage. The present study offers further experimental evidence for this assumption. Further work on the extraction properties of these common solvents toward acid salts of bases needs to be carried out.
The modified strong acid form of the Dragendorff reagent used to detect the caffeine was recommended by G. H. W. Lucas [ 11] . It was given under the name of the Ludy-Tenger reagent [ 12] , and had a concentration of bismuth subcarbonate and potassium iodide five times as strong as that given in this paper. It was found to be unsatisfactory in the original concentration. On dilution with water, it yielded a precipitate, and a heavy background on spraying. Satisfactory results were obtained with as little as 2.5 ?g of caffeine (see figure 3) with the formula given herein. After Standing for some time, the Caffeine-Dragendorff complex was found to fade. However, respraying with dilute reagent restored the colour. Kaiser & Jori [ 13] and Pohloudek-Fabini & König [ 14] have also recommended strong acid forms of the Dragendorff's reagent.
A close comparison of the IR-spectra in the three sections of figure 8 shows that the spectra of the standards and of the caffeine, heroin hydrochloride and the O 6-monoacetylmorphine-free base agree well. The major bands which are useful for identification of each of these components are given in table 5. In the heroin hydrochloride isolate from sample V-686, it should be noted that there is an extra band at 1650 cm-1 which is not present in the standard. The finger print regions in these spectra (1500 to 850 cm-1)in figure 8 compare well. The standard in the case of the heroin hydrochloride was the monohydrate, and the isolate had been dried at 105°C, which may account for any differences.
A comparison of the spectra of the O 6-monoacetylmorphine and heroin shows that both spectra have a band in the C = 0 stretching region at 1730 cm-1.Heroin has two bands (see table 5, column 5). Figure 9 illustrates the structural formulae of these acetylated alcoholic and phenolic morphine derivatives. It is concluded that the band at 1730 corresponds to the acetyl group on the O 6-alcoholic function, since paper chromatography showed no evidence of O 6-monoacetylmorphine.
One object of analysis and comparison of compositional data on different seizures from different origins is to try to establish connexions and sources of heroin found in the illicit traffic. In the case of heroin, this may be done when impurities occur.
It has been pointed out [ 1] that caffeine is one of the adulterants occasionally found in heroin in the U.S.A. However, as far as the authors know, caffeine has not been found before in heroin in the illicit traffic in Canada in the last 15 years, either in the western or eastern regions of greatest abuse of heroin.
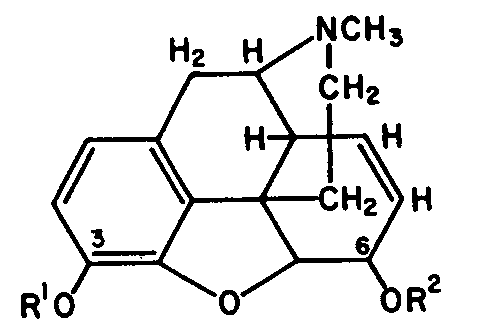
|
O3=
?
|
R1= R2=
|
H MORPHINE
|
|
O6=
?
|
R1= CH
3 CO, R2=H
|
O3- MONOACETYLMORPHINE
|
|
R1= H, R2=CH
3CO
|
O6-MONOACETYLMORPHINE
|
|
|
R1= R2= CH
3CO
|
DIACETYLMORPHINE
|
One can conclude that the caffeine-heroin mixture is unusual in our traffic.
O 66-monoacetylmorphine is said [ 1] to result from the imperfect acetylation of morphine during illicit manufacture of impure heroin. Circumstantial evidence showed our samples to have come from an illicit heroin factory outside of Macao.
The only report available to us on the composition of "crude granulated heroin" from Hong Kong is the work of Gruhzit [ 2] , which indicates it to contain 80-99% (12 samples) diacetylmorphine hydrochloride, which compares with about 65% diacetylmorphine hydrochloride and 3.5% monoacetylmorphine hydrochloride found in our samples (see table 6). No caffeine or monoacetylmorphine was indicated in Hong Kong crude granulaed heroin by any of the test data given by Gruhzit (appendix 3). Variations found in their assays were attributed to their methods and sample variations but suspected impurities were not indicated.
It is impossible therefore to draw final conclusions based on composition regarding the origin of our samples. However, very little extra work would be required to do this if samples from Hong Kong or complete analytical data were available to us. This report strongly underlines the plea made by the United Nations (3) that detailed analyses of such samples from areas where illicit heroin is thought to originate would be of value to narcotic enforcement authorities in all parts of the world.
An impure heroin has been analysed and found to be nearly 70% heroin and monoacetylmorphine, and 30% caffeine and water. It is interesting to note that the presence of about 3.5% O 6-monoacetylmorphine may be a trade-mark of the crude granulated heroin which is commonly used in "chasing the dragon" by Hong Kong addicts. Our samples were said by police to originate in Macao, and to come to Canada via Hong Kong.
The authors wish to acknowledge the helpful criticisms of Dr. R. A. Chapman, and his permission to publish this paper. We wish to thank Dr. J. Ouellet for samples, advice and assistance in carrying out preliminary experiments. Thanks are due to Mrs. Ruth Lane and Wm. Skakum for technical assistance; to G. Morris, and the biological photographic laboratories and administrative services for preparation of the MSS.
Special thanks are due to Commissioner C. W. Harvison, Royal Canadian Mounted Police, for assistance in providing the background information on the origin of the samples.
We wish to express our thanks to Mr. R. C. Hammond, the Chief of the Narcotic Control and Controlled Drugs Division, and his staff for help and continued interest in our research.
To Dr. C. A. Morrell and Dr. L. I. Pugsley our appreciation for encouragement and support is expressed.
FULTON, C. C., "Analysis of Heroin ", Bull. Narcotics, 5 , No. 2, 27-34 (1953).
002GENEST, K. & FARMILO, C. G., Bull. Narcotics, 9 , No. 4, 20-37 (1959): ibid., 10 , No. 1, 15-24 (1960).
003FARMILO, C. G . et al ., United Nations document ST/SOA/Ser. K/58 (29 Nov. 1957).
004MACDOUGALL, R. & SMITH, H. W., Royal Can. Mounted Police Seminar No. 1, 117-134 (1953).
005LOOFBOUROW, J. E. et al.,J.A.C.S., 65 , 148 (1943).
006WELSH, L. H., J. Org. Chem., 19 , 1409-1415 (1954).
007FEIGL, F., Spot Tests in Organic Analysis , 5th ed., Elsevier, N.Y., 1956.
008WRIGHT, C. R. A., J. Chem. Soc., 27 , 1031 (1874).
009WELSH, L. H., see reference 6.
010UMBERGER, C. J ., Legal Medicine Pathology and Toxicology , Appleton, Century Crofts Inc., 2nd edition, N.Y., 1954.
011LUCAS, G. H. W., Proceedings of the 7th Annual Meeting of the Candian Society of Forensic Sciences , Ottawa, 1959.
012LUDY-TENGER, F., Microchemie Ver. Mikrochimica Acta 38 , 460-465 (1952).
013KAISER, H. & JORI, H., Arch. Pharm., 287/59 , 224 (1954).
014POHLOUDEK-FABINI, R. & KÖNIG, K., Pharmazie, 13 , 13 (1958) ibid., 13 , 135 (1958).
015ANONYMOUS, "Chasing the dragon. The smoking of heroin in Hong Kong ", Bull. Narcotics, 10 , No. 3, 6, 1958.
016GRUHZIT, C. C. , ibid ., appendix 3, p. 10.
017BUTLER, W. P. & RYAN, R. L., Methods of Analysis , Pub. No. 341 (Rev. 2-60), U.S. Treasury Dept. Internal Revenue Service, Washington, Feb. 1960.
018GENEST, K. & FARMILO, C. G., J. Chromatography, 5 (1961). (In press.)
019REVITCH, E. & WEISS, G., J. of Diseases of the Nervous System, 20 , No: 5, 317 (1959).

