Experimental Biotransformation of Heroin to MAM and Morphine in vitro
Effect of Age on the Toxicity of Heroin
Results
Effect of Age on the Toxicity of Heroin
Rate of Brain Uptake of Heroin and MAM and Morphine Appearance
Discussion
Summary
Acknowledgements
Author: E. Leong WAY, M. Joseph YOUNG, John W. KEMP
Pages: 25 to 33
Creation Date: 1965/01/01
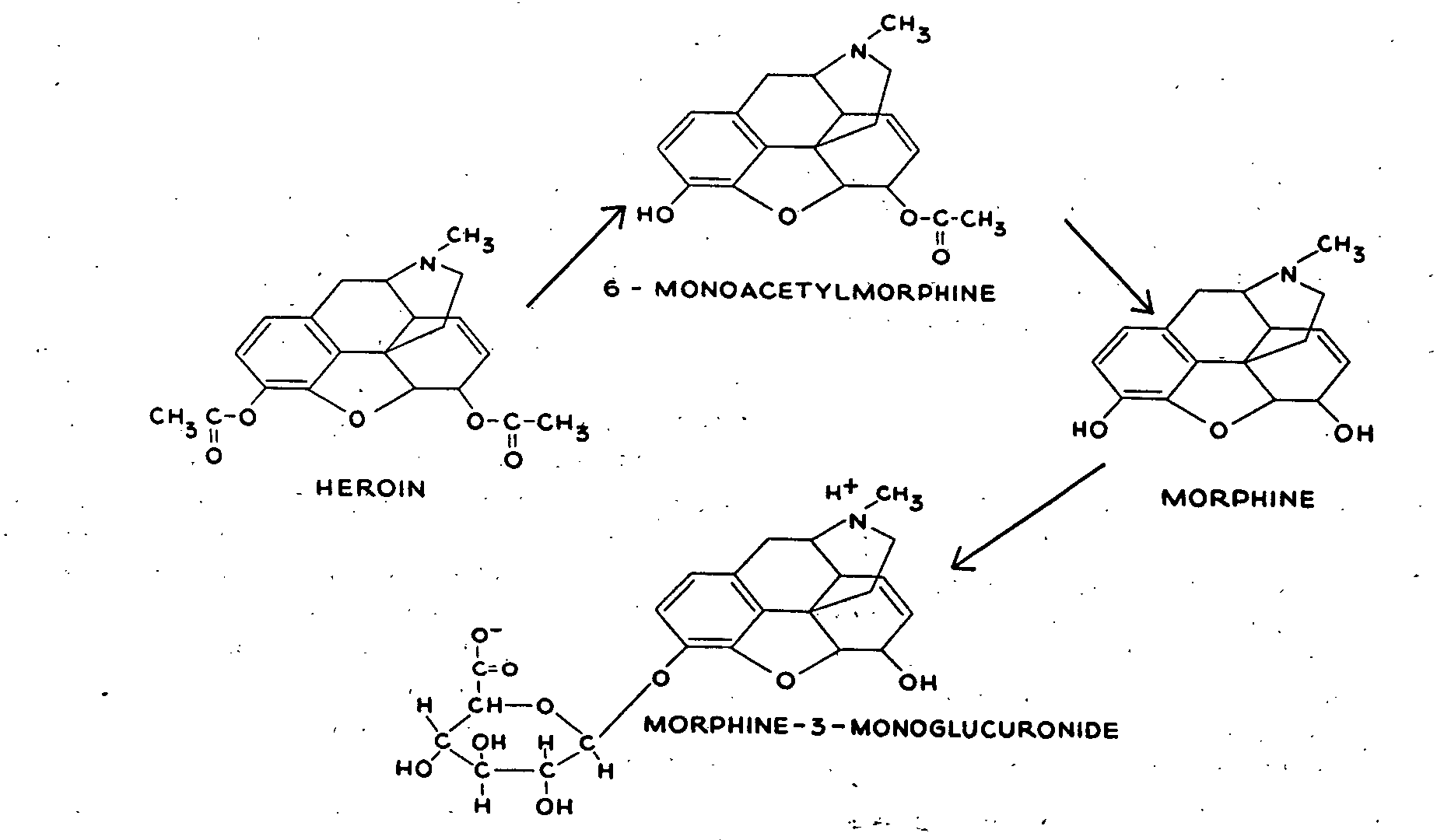
In a previous study (Way et al., 1960), we reported that heroin is rapidly hydrolyzed in the body to two products - namely, 6-monoacetylmorphine (MAM) and morphine (figure 1). The half-life for heroin appeared to be too brief for it to exert direct pharmacologic effects of any significance, excepting perhaps during a transient interval after rapid intravenous administration. We also found morphine to be more potent than heroin when the drugs were injected directly into the central nervous system, although it is generally known that heroin is more potent than morphine by the usual modes of administration. As a consequence of these findings, we proposed that the pharmacologic effects of heroin were dependent largely upon its conversion to MAM and morphine.
The above conclusion was based on experiments in mice, and it is of importance to ascertain whether such results can be part of a more general phenomenon. To this end we are reporting findings in vitro on the rat, dog and rabbit, as well as on humans and more extensive studies performed in vivo on the rat.
Various organs of several species were studied with respect to their ability to hydrolyze heroin to MAM and morphine. Comparisons were made on the brain, kidney and liver and blood from the mouse, rat, dog, rabbit, and man. Blood was diluted with nine parts of isotonic saline; the organs were similarly diluted and then homogenized in an electric-powered blender. Duplicate 10-ml aliquots were taken and incubated at 37°C with 20 μg of heroin for 20 minutes, after which the homogenates were estimated for morphine and MAM content according to a previously published method (Way et al., 1960).
Kinetic studies on heroin hydrolysis in the central nervous system of the rat were also carried out. Heroin was added to a pooled homogenate of rat brain equivalent to 20 μg of drug per gramme of wet tissue. Samples were allowed to incubate at 24°C, and at the time intervals of 2, 4, 8 and 16 minutes respectively, individual samples were analysed as above for free phenol content.
The median lethal dose of heroin was determined in 1, 2, 4, 8, 16, and 32 day old rats using freshly prepared solutions of heroin hydrochloride in concentrations which allowed injections of 0.5 ml per 20 g of body weight. Varying doses of heroin were administered intraperitoneally and the mortality was recorded for 24 hours. Rats of 8 days or younger were maintained in a ventilated oven at 30°C. Under such conditions, no deaths of animals given a saline injection occurred. The LD50 for the alkaloidal base was calculated by the method of Litchfield & Wilcoxon (1949).
Rate of Heroin Uptake and MAM and Morphine Formation by Rat Brain after Heroin Administration
Heroin hydrochloride equivalent to 75 mg/kg of the free base was injected into the popliteal space of four Sprague-Dawley rats. At fixed time-intervals of 15, 30, 60 and 120 minutes following drug administration, the animals were decapitated and their brains removed for heroin, MAM and morphine estimation.
Estimation of heroin
Heroin was determined by the methyl orange procedure for organic bases (Brodie & Udenfriend, 1945). Since MAM also reacts with methyl orange, it was necessary to correct for MAM contributions. MAM can be measured by the methyl orange procedure after separating it from heroin by countercurrent distribution in a solvent system consisting of equal parts of ethylene dichloride and 0.5 M phosphate of pH 5.5, but this procedure was less convenient to use than the following method.
Estimation of MAM and morphine
The compounds were determined by a modification of a procedure previously reported by this laboratory (Way et al., 1960). We found that the older macro method was not of sufficient sensitivity for following the compounds in the brain, and as a consequence had to devise means to detect sub-micro quantities of MAM and morphine. The micro procedure which had been developed, like the older method, is based on the alkaline extraction of the compounds with 10 % butanol in chloroform and subsequent removal from the organic solvent with an acidic buffer for estimation as free phenols with the Folin-Ciocalteu (FC) reagent. In the present procedure the sensitivity has been improved by modifying the volume and acidity of the buffer for extracting morphine and MAM from the organic solvent and by altering reagent proportions and temperature conditions for colour development with the FC reagent. Furthermore, to obtain a high degree of specificity of the method for morphine and MAM, countercurrent distribution is used to separate morphine and MAM before the final measurement is made. A brief account of the studies on the reaction condition leading to the adoption of the micro procedure is described before presenting the actual details for carrying out the procedure.
Conditions for extraction and colour development:
For the initial extraction of morphine and MAM from biologic media, the conditions of the macro procedure were easily adapted to the micro method simply by reducing the volume of 10% butanol in chloroform for extracting the drug from alkalinized homogenates of various tissues. However, in the second extraction step, where removal of the alkaloids from the organic phase is effected by acid extraction, the phosphate buffer pH 5.8, which readily extracts morphine from 10% butanol in chloroform in the macro method, was found to yield low and erratic recovery values for MAM. With a phosphate buffer of lower PH, however, consistent recoveries of MAM added to brain tissue were obtained without elevating tissue blanks. A study of the effect of varying proportions of H 3PO 4KH 2PO 4 on tissue blanks and on recoveries of 10 μg of MAM from 10% butanol in chloroform revealed that phosphate buffer at pH 1.5 would yield low brain blanks and optimal recovery of added MAM. This buffer was adopted for use in the procedure when, as expected, it was found to extract morphine readily from the organic solvent. Buffers other than phosphate of the same pH range were also studied and discarded either because recoveries were poor or tissue blanks were high.
Since the FC/carbonate ratio is important for maximal colour development and is easily influenced by many conditions (Folin & Ciocalteu, 1927), it then became necessary to assess the influence of phosphate buffer pH 1.5 on the colour index of morphine with the FC reagent. Various combinations of different concentrations of FC reagent and alkali were investigated on 5 μg quantities of drug contained in 0.5 ml. On the basis of these studies, 0.05 ml of the FC reagent (1:2) and 0.1 ml 70% K 2CO 3 were selected as a suitable combination. Although 50 or 60% K 2CO 3 probably would have been satisfactory, 70% was chosen to insure excess alkalinity for neutralizing the strongly acidic phosphate buffer. With concentrations of K 2CO 3 of 40% or less, a reaction either did not occur or was incomplete.
Finally, it was found that the colour index could be increased somewhat by heating morphine or MAM samples for one minute after addition of the FC reagent and again for three minutes after addition of 0.1 ml of 70% K 2CO 3. The tissue blank did not increase concomitantly.
Since the colorimetric method does not differentiate morphine from MAM, it is necessary to separate the compounds before estimation with FC reagent. This was accomplished by countercurrent distribution in an eight-transfer system consisting of 1 M phosphate buffer of pH 6.6 and 10% butanol in chloroform. A buffer was sought to yield partition ratios (K) for the two substances that would be roughly reciprocal to one another. After experimenting with varying proportions of 1 M KH 2PO 4 and K 2HPO 4, equal parts of each solution yielding a pH of 6.6 were found to be most satisfactory.
Procedure
Total brain free phenol: Weigh 1 g of rat tissue (wet) and homogenize it with 4 ml water. Add approximately 1 g of sodium bicarbonate and then 7.5 ml of 10% butanol in chloroform. Shake the mixture for one minute in a mechanical shaker, and centrifuge at about 3,000 rpm for 10 minutes. Aspirate the aqueous layer, add 0.6 ml of phosphate buffer, pH 1.5 to a ml aliquot of the organic layer, and shake for 2 minutes in a mechanical shaker. Centrifuge 5 minutes or until any emulsion disappears and then aspirate the organic layer. To a 0.5 ml aliquot of the buffer layer add 0.05 ml phenol reagent (FC reagent 1:2) and heat at 87°C in a water bath for one minute. Cool, add 0.1 ml 70% K 2CO 3 and heat again at the same temperature for three minutes. After cooling transfer approximately 0.3 ml of the coloured solution to a micro silica cuvette (cell size 2.5 x 10 x 25 mm., Byrocell Manufacturing Co.) and measure the optical density in the Beckman DU spectrophotometer at 675 millimicrons, using distilled water for zero setting.
Separation and estimation of MAM and morphine:
To a 6.5 ml aliquot of the organic solvent extract of brain, add 6.5 ml of 1 M K 2HPO 4-KH 2PO 4 buffer, pH 6.6. Perform an eight-plate countercurrent extraction using a mechanical rotator (about 40 rotations per minute) for mixing the two phases allowing one minute for equilibration. The bottom organic layer serves as the mobile phase. Before each transfer, centrifuge to break any emulsion. At completion of the transfers, add a drop of phenolphthalein indicator solution to the end of the eight tubes, and 16 N KOH dropwise until a faint pink appears. Add about 0.2 gramme sodium bicarbonate powder, or until the pink colour disappears and equilibrate the two liquid phases for one minute. Aspirate the top aqueous layer and mix 5 ml aliquots of the organic solvent layer from each tube with 0.6 ml phosphate buffer, pH 1.5. Take 0.5 ml aliquot from each tube and measure the free phenol present as previously described. Plot the fraction of total phenol present in each against the tube number. Fit the experimental distribution curve (Way & Bennett, 1951; Williamson & Craig, 1947) and calculate the percentage of MAM and morphine present.
Extraction loss by this procedure was found to be minimal; the recovery for both morphine and MAM after carrying through the entire procedure and correcting for aliquot loss ( ( 5/ 7.5 ) x ( 0.5 / 0.6 ) = 0.556) exceeded 90%.
The optical density for 2.5 μg of extracted drug averaged 0.145 and the deviation for a single determination was less than 0.01. Recoveries of 2.5, 5 and 10 μg of morphine or MAM added to various diluted tissues or organs (brain, 1:5; muscle, 1:10; blood 2:5) appeared to be quite satisfactory for quantitative purposes. Based on the water recovery curve, recoveries of 2.5 μg of morphine or MAM in the brain exceeded 92% and the deviation for a single determination with morphine was less than 10%. Recoveries of MAM from brain initially were more variable, but subsequently a high degree of reproducibility was obtained by diluting the organic solvent extract with two parts of fresh solvent prior to extraction with phosphate buffer.
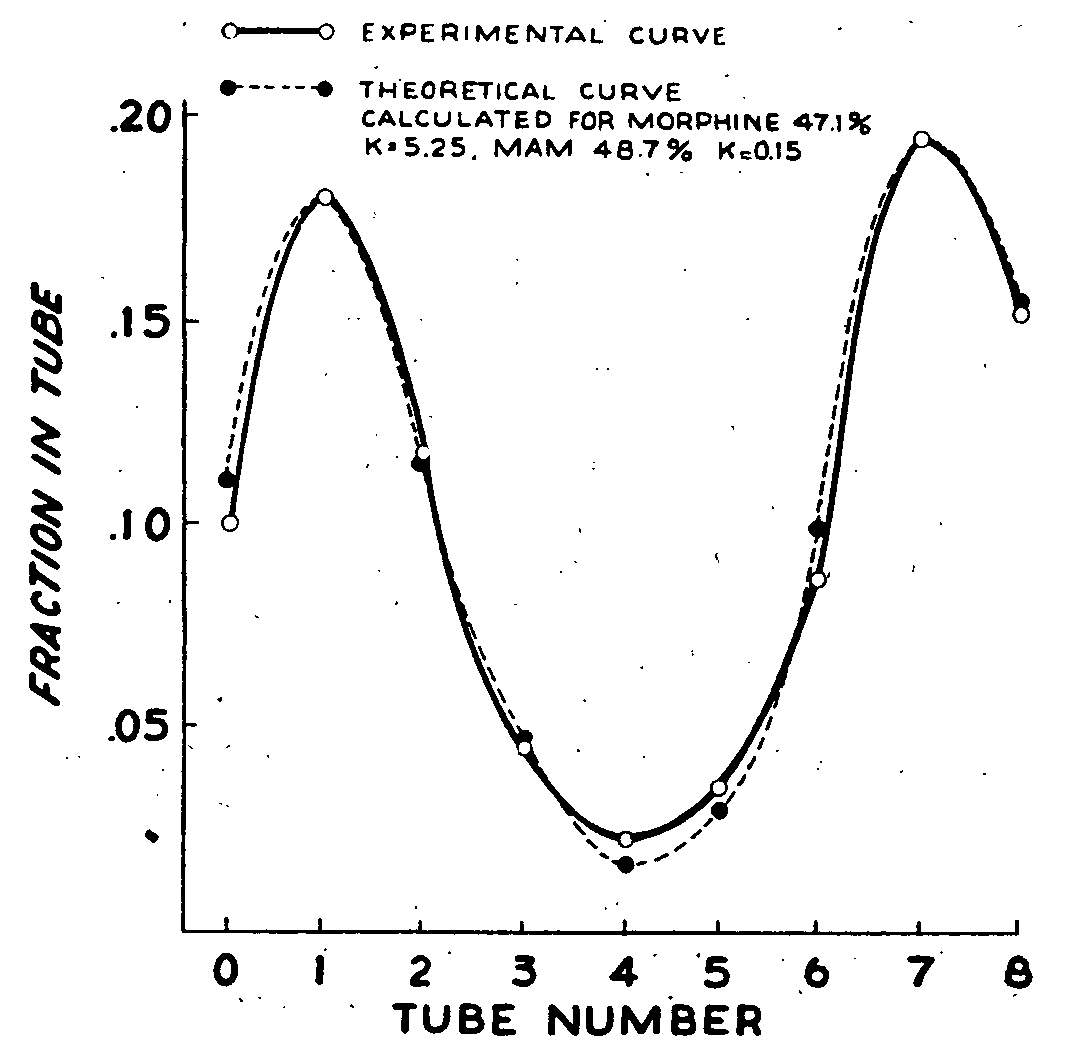
Figure 2 shows the distribution curve obtained for a mixture of morphine and MAM added to rat brain homogenate to give a 10 μg/g concentration for each substance. Two peaks which were near mirror images were obtained. The experimental curve could be nicely fitted by theoretical distribution curves for two substances with respective Ks of 5.25 and 0.15 present in equal proportions. The peak in tube 1 corresponded to a substance with the distribution characteristics for morphine and the peak in tube 7 for one resembling MAM. The K values may vary somewhat with each determination, owing probably to effects of tissue contents such as lipids or proteins or the partitioning properties of the solvent pair of the countercurrent system. This is not of major consequence since the two major peaks can still be fitted and the respective amounts of morphine and MAM present calculated.
Results Biotransformation of Heroin to MAM and Morphine in vitro
The results in general confirm previous work on the rabbit, rat and mouse (Wright, 1941, 1942; Way et al., 1960) and extend our knowledge on humans and dogs. 'The organs and tissues of all species selected for study displayed Considerable ability to metabolize heroin as evidenced by the rapid appearance of MAM and morphine (figure 3). At least 50% of the added heroin was deacetylated after 20 minutes of incubation with organ homogenates, and in many instances virtually complete hydrolysis of heroin had occurred. Under these conditions, usually more MAM than morphine was found, but in several instances, particularly with liver homogenates, there was much more morphine present. Based on studies cited immediately above, it is presumed in the present experiments that deacetylation to MAM had already occurred and the reaction had proceeded to hydrolysis of MAM to morphine.
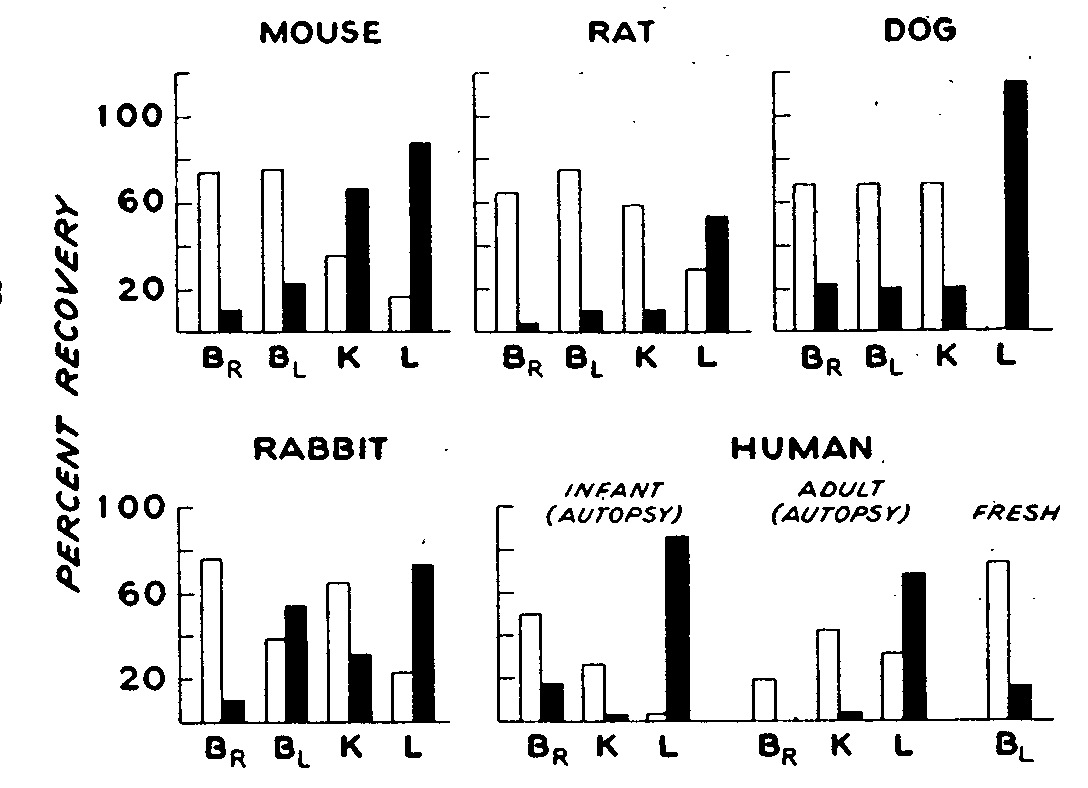
It may be noted that, although the brain was the least active of all organs studied in hydrolyzing heroin, considerable MAM and some morphine were formed by samples taken from all species. The brain of one human adult yielded minimal amounts of morphine, but brain taken from an infant was quite active. Since the age of post mortem, specimens is generally several hours, the viability of such preparations is always open to question. Undoubtedly, fresh specimens would display considerably higher and more uniform, potency.

|
Body weight a |
Surface area b |
Blood volume c |
|||||
|---|---|---|---|---|---|---|---|
|
Age in days |
Number of rats |
Grammes |
LD 50 Mg/kg |
Cm 2 |
LD 50 10+3 |
Ml |
LD 50Mg/ml |
| 1 | 25 | 6.2 |
60 (38.5- 93.6)
d
|
37.5 | 9.9 | 0.45 | 0.83 |
| 2 | 44 | 6.9 |
50 (35.2- 71.0)
|
40.0 | 8.6 | 0.48 | 0.72 |
| 4 | 36 | 9.6 |
51 (40.5- 64.3)
|
48.7 | 10.0 | 0.65 | 0.75 |
| 8 | 60 | 14.2 |
29 (21.0- 40.0)
|
61.6 | 6.7 | 0.92 | 0.45 |
| 16 | 71 | 34.6 |
115 (85.0-115.0)
|
105.2 | 37.8 | 2.2 | 1.8 |
| 32 | 30 | 94.3 |
96 (60.0-154.0)
|
191.9 | 47.2 | 5.7 | 1.6 |
a Mean values.
b Calculated from mean body weight according to Lee & Clark (1929).
c Estimated from mean body weight according to Garcia (1957).
d Figures in parentheses are 95% confidence limits.
Thus, fresh human blood was about as active as mouse, rat or dog blood in deacetylating heroin to MAM and morphine. None of these preparations, however, approached the activity of rabbit blood. The high activity of dog liver and mouse kidney should also be noted.
More extensive studies with heroin were carried out on the rat brain with respect to its rate of hydrolysis. When heroin was added to rat brain homogenate and incubated at room temperature, there was a rapid disappearance of compound as measured by the appearance of free phenol (figure 4). The latter substance represents almost entirely 6-MAM, since previous experiments with countercurrent distribution established that very little morphine is formed by this preparation under such conditions. Based on the mean values used to plot the reaction rate in figure 4, the half-life for heroin was found to be slightly greater than six minutes. A pooled homogenate of brain from several one-day-old rats was also highly active in hydrolyzing heroin, and the activity approximated that of the adult rat brain.
There was relatively little change in toxicity to heroin in the rat with increasing age. For the first eight days, the LD50 for heroin ranged between 30-60 mg/kg and then increased to about 100 mg/kg in the 16- and 32-day-old animals. The LD50 for heroin was also calculated on the basis of body surface area and blood volume, and the values are also summarized in table 1. On comparing these findings to previous results on morphine from this laboratory (Kupferberg & Way, 1963), some distinct differences are discernible. With morphine, while the LD50 remained relatively constant for the first 16 days, a sizeable increase in resistance to morphine was noted in 32-day-old animals. On a mg/kg basis the LD50 in the 32-day-old animal was roughly four times that of animals from 1 to 16 days old.
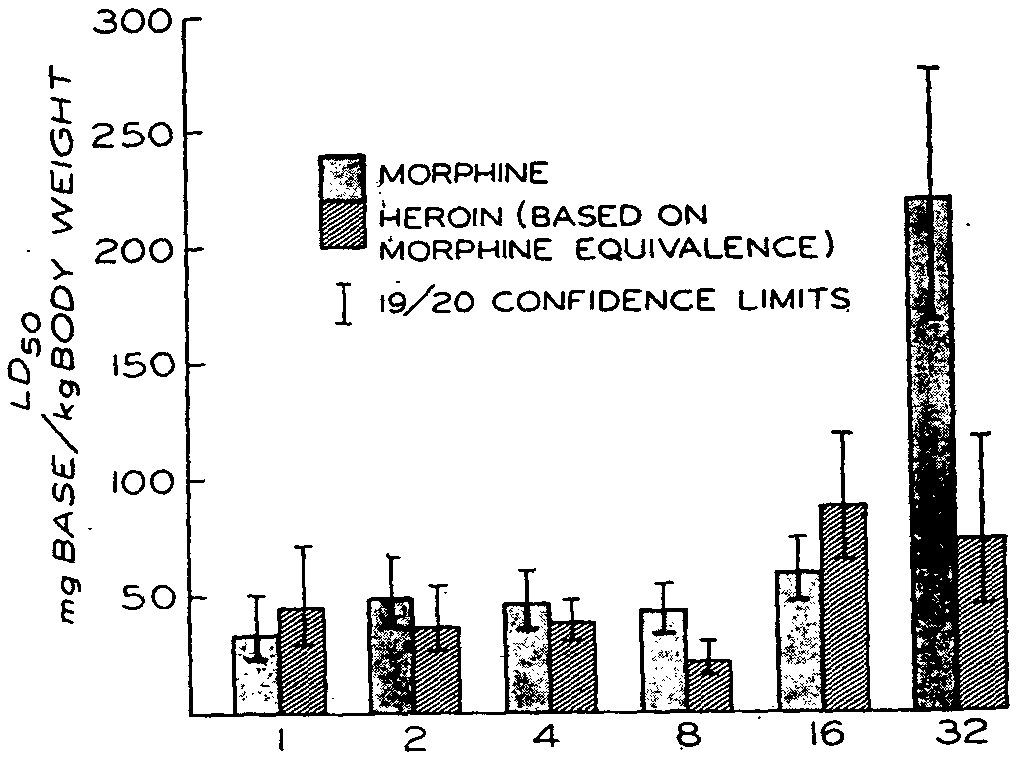
The difference in response to age between heroin and morphine is strikingly apparent on comparing the lethality of the two compounds with increasing age, as shown in figure 5. The data for heroin are taken from table 1, and have been recalculated in terms of morphine free base equivalents; the data for morphine are taken from an earlier report from this laboratory (Kupferberg & Way, 1963). It may be noted that heroin was equally toxic to morphine in the younger animals, but considerably more lethal than morphine in the older rats. The onset in effects of the two compounds did not appear to differ too much for the first 16 days. However, in the 32-day-old animals, the onset of effects for heroin appeared to be earlier than for morphine. The duration of action of both drugs with animals of varying ages appeared to be about the same with comparable doses on a weight basis.
The brain homogenates of rats injected with heroin, when extracted and analysed for MAM and morphine at various time intervals, yielded the countercurrent distribution curves shown in figure 6. Two fractions were obtained which gave a colour reaction for phenols with the FC reagent. The minor fraction with a peak in tube 1 corresponds to a substance with the distribution behaviour like morphine, and the major fraction with a peak in tube 7 to a substance with the partition characteristics of MAM. The shift in the peak of the bottom curve is due to the use of a different buffer.
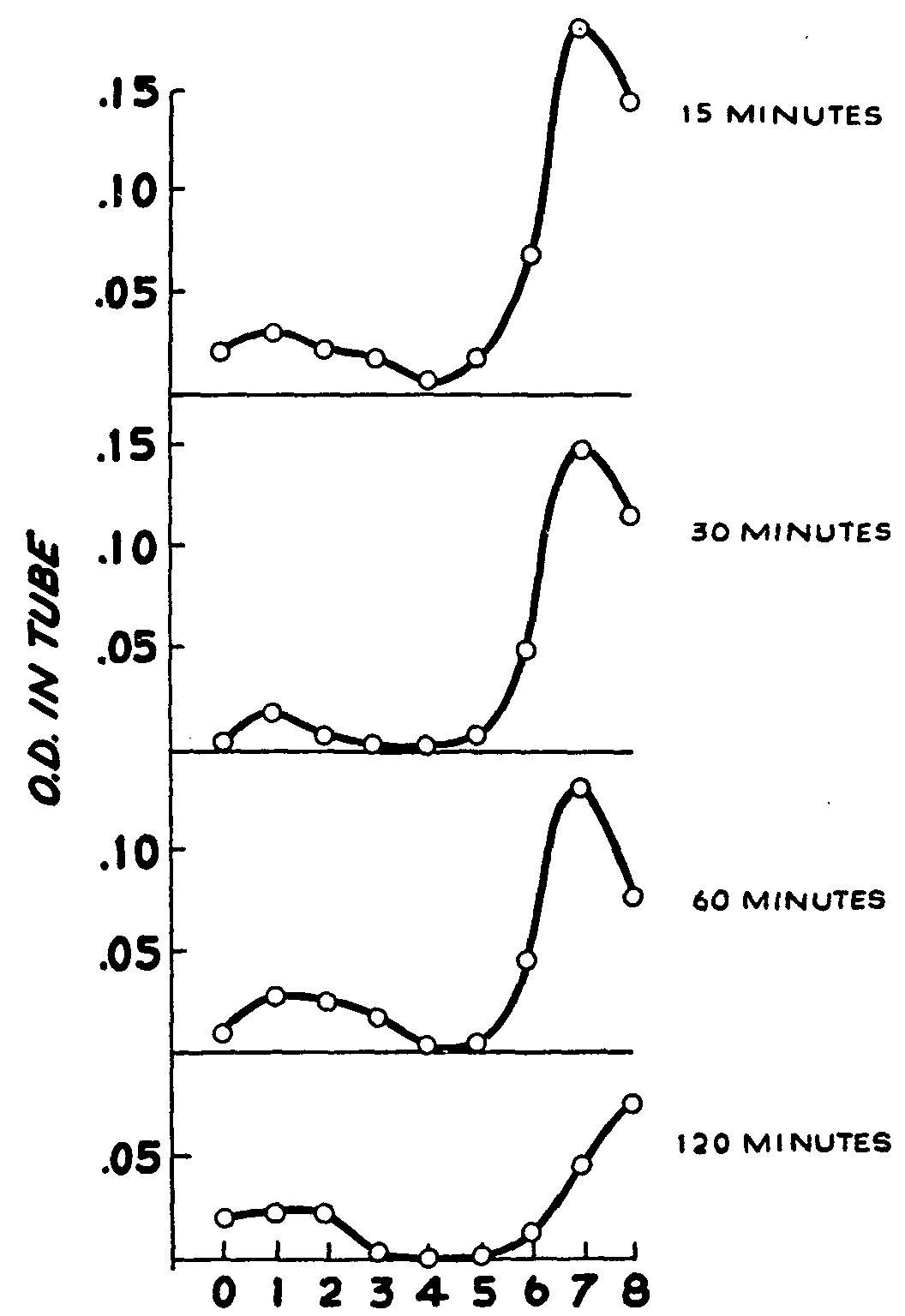
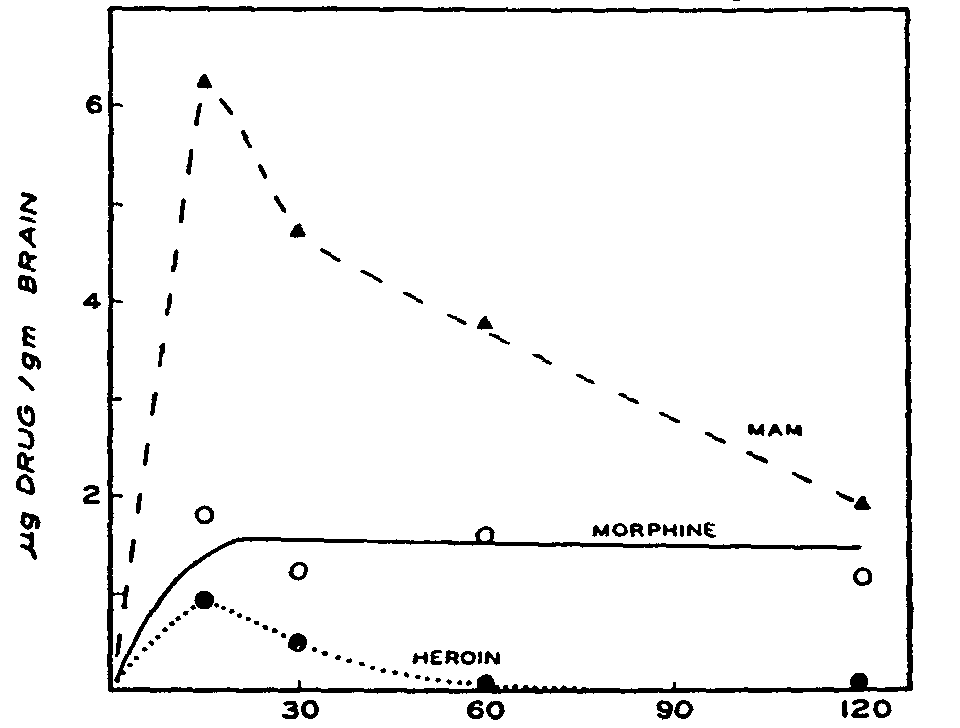
The curves were fitted and the concentrations of MAM and morphine calculated in terms of the distribution curve derived under similar conditions with the substances added to brain homogenates (figure 2). These calculated values, together with those obtained for heroin, were plotted against time to obtain the curves shown in figure 7.
Brain levels of heroin were barely detectable. As shown in figure 7, at 15 minutes a level of 0.9 μg per g of brain tissue was detected, which fell to 0.5 μg per g at 30 minutes, and to less than this amount at the 1- and 2-hour periods. Concomitantly, there was a peak level of 6.3 μg per g of MAM at 15 minutes, followed by a fairly rapid decline to 1.9 μg per g at 2 hours, but this level was still more than double the highest concentration attained by heroin. Thus, both heroin and MAM appeared early in the brain, highest levels of the drug being attained at the first sampling time of 15 minutes, and by 30 minutes the levels for both compounds were declining. On the other hand, the morphine content of the brain remained relatively constant around 1.5 μg per g throughout the experimental period. At the last sampling period (2 hours), the animals still appeared subjectively to be under drug effects.
The results further establish that heroin is rapidly metabolized to MAM and morphine, and furnish additional evidence to support the postulate that the major pharmacologic effects of heroin are dependent upon MAM and morphine formation (Way et al., 1960).
We are not the first to suggest that the actions of heroin are dependent on its hydrolytic products. In 1935, two groups of investigators (Eddy & Howes, and Wright & Barbour) independently suggested that heroin may act principally as MAM because the two compounds were found to be about equivalent in their pharmacologic effects. Wright later suggested, in 1942, on the basis of in vitro studies, that heroin might be acting as morphine. Our present and previous study represent the initial attempt to correlate pharmacologic effects with brain levels of the three compounds in question. Our evidence indicates that the pharmacologic actions of heroin are indeed dependent largely upon its deacetylation to MAM and then to morphine. The biologic half-life of heroin is so brief that, other than perhaps an initial transient effect, the activity of the drug must of certainty reside in one or both of its two chief bio-transformation products.
The experiments in vitro suggest that the ability to hydrolyze heroin rapidly is a phenomenon shared by many mammals, and it appears, therefore, that the metabolic pathway for heroin in many species is very similar. Incubates of the liver, kidney, blood and brain of the mouse, rat, rabbit, dog and human are highly active in deacetylating heroin at both the 3-carbon and the 6-carbon positions. The 3-acetyl is particularly labile to hydrolysis with resultant MAM formation. Hydrolysis of MAM at the 6-acetyl position to morphine, while slower, still occurs fairly rapidly. The liver is the most active organ for both reactions but even with rat brain homogenates the mean half-life for MAM formation at room temperature was found to be only about six minutes.
The above results support earlier manometric work where hydrolysis of heroin was demonstrated indirectly without identification or measurement of the MAM and morphine that presumably was formed (Wright, 1941, 1942; Ellis; 1948). According to these investigators, who studied the nature of the enzyme(s) involved in the reaction, there may be two classes of enzymes, and the enzymes are not esterases which hydrolyze acetyl-choline, acetylsalicylic acid or methylbutyrate but appear to be tributyrinase.
The rapidity of the hydrolysis of heroin to MAM and then to morphine in vitro suggests that it would be highly unlikely for heroin to exert direct pharmacologic effects for any significant duration and hence the biotransformation products, MAM and/or morphine, might be expected to contribute considerably to the over-all pharmacologic effects of heroin.
In the toxicity experiments, the relatively little change in the lethality of heroin with increasing age, in contrast to the marked decrease obtained with morphine, supports the postulate that the pharmacologic effects of heroin are largely dependent upon morphine formation and suggests that heroin and its chief metabolite, MAM, function primarily as carriers to facilitate morphine availability at receptor sites in the central nervous system. Since a previous study in this laboratory (Kupferberg & Way, 1963) has established that the decrease sensitivity to morphine in the rat with increasing age is attributable primarily to the development of a blood-brain barrier to morphine, one would not expect this factor to be important for heroin. If the barrier is relative and related to diffusion rates, heroin, by virtue of its greater lipoid solubility than morphine, should gain ready access to the CNS of the adult rat, and, hence, there should be little change in apparent potency of the drug with increasing age. Furthermore, if no blood-brain barrier to morphine exists early in life and if toxic effects of heroin are dependent upon morphine formation, then depending upon the rate of this conversion, heroin can be equi-toxic or less toxic than morphine but not more so. Only when the barrier to morphine develops should heroin become more potent than morphine.
The results are consistent with these expectations. Whereas the LD50 for morphine increased to roughly fourfold to fivefold from the day of birth to 32 days of age, the LD50 increase for heroin over the same period was but 1.3 times. Moreover, the LD50 for heroin for the first 16 days was about equivalent to that for morphine, but by 32 days this order was markedly altered presumably because of the development of the brain-barrier to morphine.
Finally, the brain uptake studies on heroin indicate a very rapid disappearance of heroin from the brain and the sojourn of the compound at this site does not appear consistent with its duration of action. The decay curve for MAM in the brain indicates that MAM can make a significant contribution to the sum total of heroin effects but its rate of decay suggests that any contributions it may make should be during the first two hours after heroin administration. Since the pharmacologic effects noted persisted several hours beyond this period, it appears more reasonable to ascribe the later effects to morphine. Owing to limitations in methodology, it was not possible to assess quantitatively the relative pharmacologic contributions of MAM and morphine for the initial two hours after heroin administration.
The level of 1-2 μg morphine/g of brain following heroin administration is in excess of that attainable by a comparable dose of morphine. When a rat was injected with 75 mg/kg of morphine intrapopliteally, the brain level of morphine at 15 minutes was found to be but 0.9 μg/g. Moreover, Woods (1954) has shown that brain morphine levels in the rat at various times after 150 mg/kg subcutaneously were less than 0.5 μg/g. Clearly then, morphine must have difficulty traversing the blood-brain barrier, and heroin and MAM can function as carriers to enhance morphine access to the brain. This barrier to morphine, however, is relative since 75 mg/kg of morphine by rapid intravenous administration was found to yield a level in the brain slightly in excess of 5 μg/g.
The levels of morphine found in the CNS after heroin administration are more than enough to elicit the classic effects of morphine. Brain levels of morphine at pharmacologic doses (2 mg/kg) range less than 0.1 μg/g (Adler, Elliott & George, 1957). Thus, the morphine levels we noted represent toxic levels and this was evinced by the fact that animals receiving the heroin became cataleptic within two minutes and remained so for at least two hours. Even after several hours, animals were visibly under drug effects. Obviously an extremely sensitive response to morphine is exhibited by the central nervous system and this selectivity of action argues in favour of our hypothesis.
It should be further pointed out that these findings on the rat are analogous to previous results on the mouse (Way et al., 1960), and in the previous study we were also able to demonstrate that morphine is more toxic than heroin when the compounds are injected intracerebrally.
Recent studies by others may also be cited to support our hypothesis. Clinical studies by Lasagna and associates (1955) indicate that addicts are unable to distinguish between heroin and morphine when the drugs were administered subcutaneously. When administered intravenously, doses of these drugs had comparable action time courses, and there was no marked differences in their ability to produce feeling of " euphoria ", ambition, nervousness, relaxation, drowsiness or sleepiness. Furthermore, the rates of tolerance development to heroin and morphine were roughly the same (Fraser et al., 1961; Martin & Fraser, 1961). Thus, it appears likely that heroin per se has very little direct pharmacologic actions and most of its effects follow its conversion to MAM and morphine.
At the present writing, it is not possible to assess more quantitatively the temporal aspects with respect to the relative contributions of MAM and morphine to the pharmacologic effects of heroin. It is hoped that a projected exploration of these relationships with labelled isotopes of these compounds will be profitable.
Homogenates of the liver, kidney, brain and blood from the mouse, rat, rabbit, dog and man were highly active in hydrolyzing heroin to 6-monoacetylmorphine (MAM) and to a lesser degree in hydrolyzing MAM to morphine. The half-life for the hydrolysis of heroin by rat brain homogenate at room temperature was found to be approximately 6 minutes.
In rats which develop a brain barrier to morphine with increasing age, the toxicity of heroin in contrast to morphine changed relatively little with increasing age. Whereas the increase in LD50 with morphine from animals at birth to 32 days increased about five-fold, the increase with heroin was found to be less than twofold. The onset of pharmacologic effect with the two compounds differed little early in life but as the animals matured the onset of morphine effects appeared to become slower than those of heroin.
Brain levels of heroin in rats injected intrapopliteally with the compound appeared as early as 15 minutes, and the amount present declined rapidly thereafter. Parallel with the rapid disappearance of heroin was the rapid appearance of MAM in the brain. In contrast, morphine levels appeared more slowly and were more sustained. Of the three substances present in the brain, morphine appeared to correlate best with the duration of toxic effects.
On the basis of the findings, it was proposed that unchanged heroin can exert only minor transient pharmacologic effects and the chief actions of the compound are the consequence of its biotransformation to MAM and morphine. It is postulated further that morphine is probably responsible for most of the pharmacologic effects of heroin, and that heroin and MAM function largely as carriers to facilitate access of morphine to its receptor sites in the central nervous system.
This study was supported in part by research grant GM-01839 from the National Institutes of Health, U.S. Public Health Service. It is a pleasure to acknowledge the advice of Dr. T. K. Adler and the capable technical assistance of Mr. Mogens Lauesen.
Adler, T. K., Elliott, H. W. & George, R. (1957), J. Pharmacol., 120 , 475.
Brodie, B. B. & Udenfriend, S. (1945), J. biol. Chem., 158, 705.
Craig, L. C., Columbic, K., Mighton, H. & Titus, E. (1945), J. biol. Chem., 161, 321.
Eddy, N. B. & Howes, H. A. (1935), J. Pharmacol., 141, 105.
Ellis, S. (1948), J. Pharmacol., 94, 130.
Folin, O. & Ciocalteu, V. (1927), J. biol. Chem. 73, 627.
Fraser, H. F., Van horn, G. D., Martin, W. R., Wolfbach, A. B. & Isbell, H. (1961), J. Pharmacol., 133, 271.
Garcia, J. F. (1957), Amer. J. Physiol., 190, 19.
Kupferberg, H. & Way, E. Leong (1963), J. Pharmacol., 141, 105.
Lasagna, L., Felsinger, J. M. Von & Beecher, H. K. (1955), J. Amer. Med. Ass'n, 157, 1006.
Lee, M. O. & Clark, E. (1929), Amer. J. Physiol., 89, 24.
Litchfield, J. T., Jr. & Wilcoxon, F. (1949), J. Pharmacol., 96, 99.
Martin, W. R. & Fraser, H. F. (1961), J. Pharmacol., 133, 388.
Way, E. Leong, & Bennet, B. M. (1951), J. biol. chem., 192, 335.
Way, E. Leong, Kemp, J. W., Young, J. M. & Grassetti, D. R. (1960), J. Pharmacol. 129, 144-154.
Williamson, B. & Craig, L. C. (1947), J. biol. Chem., 173, 63.
Woods, L. A. (1945), J. Pharmacol., 112, 158.
Wright, C. I. (1941), J. Pharmacol, 71, 164.
Wright, C. I. (1942), J. Pharmacol, 75, 328.
Wright, C. I. & Barbour, F. A. (1935), J. Pharmacol., 54, 25.

