Summary
Table of contents
1. Determination of total ecgonine in coca paste
2. Determination of cocaine in coca paste
Chromatographic, spectrophotometric and polarimetric investigations of cocaine and the products of cocaine hydrolysis
Legend
4. Application of the results obtained to the analysis of residues from the manufacture of cocaine from coca paste
Bibliography
Author: Francesco TOFFOLI , Ustik AVICO
Pages: 27 to 36
Creation Date: 1965/01/01
The methods of determining "total ecgonine" are discussed; a method is described for determining the cocaine in coca paste (or raw cocaine): low-temperature oxidation of the secondary alkaloids with permanganate, extraction with ether, and weighing of the cocaine base.
The absorption spectra are specified, and the specific optical rotations at various pH-values are given for ecgonine and anhydroecgonine, from which rotations the apparent constants of acid and basic dissociation have been deduced.
The rotations obtained were measured by comparison with those obtained in the acid hydrolysis of cocaine.
A description is given of a method of paper chromatography by which the ecgonine and anhydroecgonine in the mixtures are separated and then determined, and the results are given of the determinations of these two components, and of the methyl ester of anhydroecgonine, in the residues from the manufacture of cocaine from coca paste.
Determination of total ecgonine in coca paste.
Determination of cocaine in coca paste.
Chromatographic, spectrophotometric and polarimetric investigations of cocaine and the products of cocaine hydrolysis:
preparation of ecgonine;
preparation of anhydroecgonine;
paper chromatography of mixtures of ecgonine and anhydroecgonine;
ultraviolet spectra of ecgonine and anhydroecgonine;
polarimetric measurements of solutions of ecgonine and anhydroecgonine at various pH-values and deduction of the constants of acidic and basic dissociation;
polarimetric measurements of liquids yielded by the acid hydrolysis of cocaine.
Application of the results obtained to the analysis of residues from the manufacture of cocaine from coca paste.
Both coca leaf and coca paste (or raw cocaine) are imported into Italy. Coca paste is a raw base, with a cocaine content usually in excess of 70 per cent, extracted from the leaves in the country of origin. While the leaves are chiefly used for liquid preparations, cocaine is mainly extracted from coca paste by a method which, in its general lines, derives from that proposed by Chemnitius [ (11)] [ (15)] .
The method consists in the oxidation of the secondary alkaloids with permanganate in an acetic acid or sulphuric acid medium, precipitation of the base with alkali, and extraction and dissolution in a suitable solvent from which the hydrochloride is precipitated by treatment with hydrochloric acid.
For the controls which, for the purpose of combating the illicit traffic in drugs, the authorities exercise on the importation of coca paste and on the production of cocaine it is important to know: ( a) the quantity of cocaine present as such in the coca paste; ( b) the aggregate quantity of ecgonine alkaloids (or total ecgonine), since it would also be possible to prepare further cocaine by hydrolysis and subsequent partial synthesis from the ecgonine alkaloids differing from cocaine; and ( c) the nature and quantity of the substances which accumulate in the residues from the manufacture of cocaine from coca paste; since these data make it possible to reconstruct the balance of manufacture, to justify losses, and possibly to improve the process of extraction.
Many methods are known for determining the total ecgonine in coca leaf or coca paste. They are based:
On measurement of the optical rotation of the liquid obtained by acid hydrolysis of the alkaloids [ (2)] , [ (9)] , [ (13)] , [ (14)] , [ (23)] ;
On titration of the aromatic acids which are formed by alkaline hydrolysis in an aqueous acetone medium and which are separated by successive acidification and extraction [ (13)] [ (14)] ;
On determination by weight and volume of the ecgonine obtained in the form of ecgonine hydrochloride residue after hydrolysis [ (4)] , [ (5)] , [ (17)] ;
On the volumetric determination of the alkaloids extractable with petroleum ether or a mixture of ethyl ether and petroleum ether [ (13)] , [ (14)] , [ (20)] .
The methods referred to are not all of equal value; they exhibit merits and defects which will be briefly considered here.
The "( a) " methods. On consulting the voluminous literature on the subject, we experienced some difficulty in interpreting the optical rotations, and we have thought it advisable, so as to restore some degree of orderliness, to introduce the molecular rotation [M] also:
|
[M]=
|
M
|
[α]
|
| 100 |
in which M is the molecular weight of the substance whose specific rotation [α∝] has been determined. The molecular rotation so defined (21) represents the rotation which would be observed in a molar solution at the thickness of 1 metre, assuming the rotation to be proportional to the concentration. The use of this magnitude makes it easier to compare the rotatory powers of substances belonging to the same series with those of the same substance in differing conditions.
A. W. K. de Jong (9) hydrolizes the alkaloid fraction, extracted from 50 g of leaf, by boiling for one hour with 15 ml of 2-N HC1, separates the acids by filtration, makes up the volume of the aqueous acid solution to 25 ml, decolorizes with animal charcoal, measures the optical rotation, and expresses the result, as ecgonine hydrochloride contained in the 25 ml, by the formula:
|
C =
|
25 |
α
|
| 57 |
De Jong expressly asserts ( loc. cit., page 988) that the figure -57.0°for the empirical specific rotation of ecgo-nine hydrochloride in a solution of 1-N to 2-N HC1, which he introduces into the formula set out above, is that quoted by Einhorn (2), which he himself confirmed by working on the acid-hydrolysis liquids obtained from cocaine base, cocaine hydrochloride and benzoylecgo-nine, the mean value he determined being -56.1°±1.3.
Thus, in de Jong's method, the value -57.0°([M] D = -126.0°), referred to ecgonine hydrochloride and obtained empirically on liquids from cocaine hydrolysis carried out with 2-N HC1, is accepted.
According to J. R. Nicholls (13) also, the value of the empirical specific rotation in those conditions is -57.0°.
From the fact that both authors give the same value (-57.0°) it might be thought that both had reached the same result. In our view, however, the identity of the figures is a coincidence, the same figure having a different meaning for the two authors: ( a) for de Jong, who continued the boiling for only one hour (with the result that hydrolysis was, as we shall see, incomplete), it means the specific rotatory power referred to ecgonine hydrochloride of molecular weight 221.7; ( b) for Nicholls, who continued the boiling for five hours (with the result that hydrolysis was complete), it means the specific rotatory power referred to ecgonine (anhydrous) of molecular weight 185.2.
By boiling for one hour in 2-N HC1, de Jong does not achieve complete conversion of the cocaine into a mixture of ecgonine and anhydroecgonine, but obtains a mixture of products having a higher molecular rotatory power. He fails to make sure that hydrolysis is complete either by extracting the basic products (cocaine, methyl ecgonine, methyl anhydroecgonine) or by prolonging hydrolysis until the results obtained no longer change with continued boiling.
Our experiments, however, do not confirm the values obtained by de Jong; it is in fact understandable that, in slightly different conditions, incomplete hydrolysis might yield substantially different results. They do, however, fully confirm Nicholls's results and therefore his method.
In table 1 we have set forth the specific rotation [α] D supplied by the various authors. The molecular weights, and therefore, the corresponding molecular rotations [M] D, are scarcely ever explicitly supplied by the authors; they have been supplied by us on the basis of the context to enable the comparisons to be made.
The "( b)" methods. These are used to check method ( a) with regard to the cocaine, cinnamyl-cocaine and truxillins, but they do not take account of the possible presence in the coca paste of methyl ecgonine, which does not contain esterified acids to the secondary alcoholic function.
The "( c)" methods. The original Greshof method has the drawback of requiring laborious correction - which moreover is not accurate - for the mineral substances (ashes) present in the coca paste (17).
The" ( d)" methods. The alkaloids which are extracted with ethyl ether or petroleum ether or mixtures may also contain tropinic not ecgoninic alkaloids or they may not contain any fraction of ecgoninic alkaloids.
A modification of the Nicholls (13) and Gilbert method has recently been proposed, in which the mixture of ethyl ether and petroleum ether is replaced by petroleum ether in the extraction of the coca paste alkaloids. According to the author (20) the modification yields a result 7 per cent lower than that provided by the Nicholls (a) and (b) methods
|
Substance |
Molecular weight |
[α] D |
[M] D |
Solvent |
Author |
|---|---|---|---|---|---|
|
Ecgonine
|
203.2 |
-45.6°
|
-92.7°
|
H
2O
|
A.W.K. de Jong (9), p. 995
|
|
Ecgonine
|
203.2 |
-45.2°
|
-91.0°
|
H
2O
|
M.R. Bell and S. Archer (23)
|
|
Ecgonine
|
203.2 |
-45°
|
-91.4°
|
H
2O
|
Hesse (3)
|
|
Ecgonine
|
185.2 |
-57.0°
|
-105.5°
|
HCl.2N
|
J. R. Nicholls (13), p. 158
|
|
Ecgonine hydrochloride
|
221.7 |
-56.1°
|
-124.2°
|
HCl.1N
|
De Jong (9), p. 988
|
|
Ecgonine hydrochloride
|
221.7 |
-48.9°
|
-108.2°
|
HCl.2N
|
De Jong (9), p. 988
|
|
Ecgonine hydrochloride
|
221.7 |
-57.0°
|
-126.2°
|
H
2O
|
A. Einhorn (2)
|
|
Ecgonine hydrochloride
|
-
|
-47.1°
|
-104.2°
|
?
|
De Jong (9), p. 995
|
|
Ecgonine hydrochloride
|
221.7 |
- 59.0°
|
- 130.8°
|
H
2O
|
Herrera Orosco
|
|
Anhydroecgonine
|
167.2 |
-84.6°
|
-141.5°
|
H
2O
|
De Jong (9), p. 996
|
|
Anhydroecgonine hydrochloride
|
203.7 |
-72.4°
|
- 147.2°
|
H
2O
|
De Jong (9), p. 997
|
|
Anhydroecgonine hydrochloride
|
-
|
-62.0°*
|
-126.2°
|
H
2O
|
De Jong (9), p. 997
|
|
Anhydroecgonine hydrochloride H
2O
|
221.7 |
-66.0°
|
-146.2°
|
H
2O
|
De Jong (9), p. 997
|
|
Anhydroecgonine hydrochloride H
2O
|
221.7 |
-61.5°
|
- 136.1°
|
H
2O
|
A. Einhorn (2)
|
|
Anhydroecgonine hydrochloride H
2O
|
221.7 |
-62°
|
-137.2°
|
?
|
Hesse, quoted by de Jong, p. 997
|
|
Anhydroecgonine hydrochloride H
2O
|
221.7 |
-56.6°
*
|
-123.3°
|
H
2O
|
De Jong (9), p. 997
|
After mutarotation.
The change may be of some utility in conjunction with the other methods for evaluating the total ecgonine in coca paste, but it does not seem to us that the results obtained with this modification should be preferred to those yielded by the methods proposed by Nicholls, because the petroleum ether does not extract the truxillins [ (1)] , [ (8)] , [ (19)] , which nevertheless are ecgonine alkaloids.
With regard, however, to the determination of the total ecgonine in coca paste, while the known methods exhibit defects when taken individually, they can be regarded as satisfactory taken as a whole.
However, no simple method has been described for determining the cocaine. Cocaine determination is more important than determination of total ecgonine, because with the present industrial methods it is mainly the cocaine present as such that is extracted with high yields, while the other ecgoninic alkaloids, normally present in small quantities in coca paste, are either not used at all or, if they are used for preparing further cocaine, give very low yields and are of doubtful economic advantage for the purposes of the licit trade.
The method proposed here for determining the cocaine in coca paste derives from the industrial method of Chemnitius (11) and from the Nicholls method ofseparation and dosage of the cocaine in mixtures with novocaine [ (13)] .
We have confirmed experimentally that, in the conditions chosen, the cocaine is not acted upon by the permanganate, and accordingly reaches the final residue without loss.
5-1.0 gramme of coca paste is dissolved in 10 ml of 3-per-cent sulphuric acid cooled to 0 °C. This temperature being maintained, and with frequent shaking, 8 ml of a solution containing 6 per cent potassium permanganate' and 10 per cent sulphuric acid is added in successive fractions of 1 ml, the solution being left to stand for 5-10 minutes to decolorize after each addition before the next addition is made. After the last addition the solution is left to stand for half an hour at 0 °C.
Finely-ground oxalic acid is added until the precipitate which has formed has completely dissolved; this leaves a clear and colourless solution. Extraction is performed twice with ethyl ether, which is discarded. The solution is alkalized with ammonia and extraction is performed four times, each time with a volume of ethyl ether equal to the volume of the aqueous phase. Losses being avoided, the ethyl ether is dried with sodium sulfate and evaporated to dryness in a weighed flask. The flask is first placed in an oven at 60 °C for two hours and then in a sulphuric acid dessicator for twelve hours, and weighed.
The residue is dissolved in 20 ml absolute alcohol, 20 ml water is added, and the whole is weighed. From the total weight of the solution (difference between the weight last determined and the weight of the empty flask) divided by 0.926 (density of the solution at 20 °C) the volume of the liquid is determined for the purposes of computing the cocaine concentration from the polarimetric reading.
In these conditions, i.e. in alcohol of about 54°, it is possible to assume for the cocaine base (molecular weight 303.35) the specific rotation [α] D = -35-0° ± 0.5°; [M] D = - 106°reported in the literature (5) and confirmed by us.
With a portion of this liquid diluted 1 : 1,000 with a decinormal solution of HCl the ultraviolet absorption at 274 mµ is determined.
In decinormal HCl solution it can be assumed that the molar extinction for cocaine at 274 mµ will be ε = 1.130 ± 60, and therefore E (1% /1cm) = 38.2 ± 2.0.
From our measurements the molecular extinction values reported earlier were determined for the two maxima and for the minimum of ultraviolet absorption in decinormal HCl solution.
However, the safest method of determination consists in weighing the residue; the other methods are open to major sources of error.
The result of the oxidations carried out with permanganate on several samples of coca paste was checked by chromatographic and electrophoretic examinations.
The chromatographic method described by L. Leiserson and T. B. Walker (18) for the separation of tobacco alkaloids was used, this method having given us good results in the separation of the alkaloids of Lupinus albus (unpublished research). The coca alkaloids, however, being less soluble in water than tobacco alkaloids, advanced with the leading face of the solvent, and their separation was not achieved. This drawback was overcome by increasing the proportion of acetic acid in the buffering aqueous mixture used to saturate the n-butyl alcohol and by eliminating the buffering of the paper, which was done with the same aqueous solution of sodium acetate and acetic acid.
The chromatographic method ultimately adopted is summarized below.
500 ml n-butyl alcohol is saturated by being shaken for 15 minutes in a solution of 24 g trihydro acetate and 15 ml glacial acetic acid in 1 litre of water. The alcoholic layer is placed in the trough with the prepared paper, and after an interval of 24 hours the paper is submerged to a depth of 2 mm, followed by ascending chromatography. After 24 hours, the papers are washed, dried, examined first in the ultraviolet light provided by a Philips germicidal lamp equipped with a Corning 7910 filter of 70% 245 .mμ radiation, and then developed withDragendorff reagent diluted with acetic acid in the proportion of 1:4.
The chromatographs reveal in. the coca paste a very strong stain with R F = 0.53, a less strong stain with R F = 0.64, and a weak stain with R F = 0.93. Only the first stain remains in the product which has undergone oxidation; the other two disappear entirely.
Electrophoresis was carried out as follows: 0.2 mg of coca paste, or the oxidation residue in the form of 10 μl either of aqueous acid solution (HC1 N/10) or of aqueous alcoholic solution, is placed on a strip of paper 25 cm long which is soaked with a solution of phosphates of pH 7.08 ± 0.05 and subjected to a potential difference of 250 V (10 V/cm).
With the coca paste a larger stain is obtained which, in the conditions described, moves towards the cathode, and a much smaller stain which does not move; with the product of oxidation, only the first stain remains unchanged, while the second disappears completely.
The methods at present used for evaluating the residues from the processing of coca paste and based essentially on the determination of optical rotation * presuppose that these residues contain, as a secondary product of the ecgonine alkaloids, only ecgonine or a mixture of ecgonine and anhydroecgonine in constant proportions.
The present investigation shows, however, that the composition of the ecgonine alkaloids in nitrogenous derivatives is more complex and is variable; for this reason, separate determination of the principal nitrogenous compounds cannot be omitted in the evaluation of the residues in question.
First of all we proposed to separate the ecgonine from the anhydroecgonine and determine the quantity of each in the presence of the other.
In preparing the pure products were observed that, despite the repeated crystallizations, the ecgonine obtained by acid hydrolysis (with HC1 2N) from cocaine was always heavily contaminated with anhydroecgonine. This impurity has a particularly marked influence on the absorption of ultraviolet radiation and on the rotatory power; for in the absorption spectra provided by Farmilo (16), Castille and Ruppol (12), and Herrero Orosco a degree of absorption is observable that does not seem to be warranted by the molecular structure of ecgonine.
Information kindly supplied by Messrs. SIMES, Milan.
|
Original sample |
Basic fraction |
Neutral aqueous fraction (alcoholic extract) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
| 46 |
Liquid with lumps
|
5.3 | 1.1 | 30.0 | 0.965 | 0.914 |
0.29/0.71
|
3.7 | 8.1 |
| 47 |
Semi-solid
|
6.9 | 3.7 | 50.0 | 2.42 | 2.48 |
0.20/0.80
|
6.4 | 23.0 |
| 48 |
Dry lumps
|
1.4 |
-
|
45.5 | 2.12 | 1.88 |
0.24/0.76
|
6.7 | 19.1 |
| 49 |
Liquid with lumps
|
3.6 |
-
|
55.7 | 1.90 | 1.88 |
0.22/0.78
|
5.5 | 17.6 |
| 50 |
Homogeneous liquid
|
2.6 |
-
|
21.5 | 0.68 | 0.69 |
0.46/0.54
|
4.1 | 4.4 |
| 51 |
Homogeneous liquid
|
11.2 | 3.9 | 24.8 | 0.78 | 0.69 |
0.39/0.61
|
4.0 | 5.6 |
All the figures are given in grammes and referred to 100 g of the original sample examined, undried.
Number of sample
Consistency
Weight of the basic fraction extracted with ether
Methyl ecgonine anhydride in the aforesaid basic fraction computed from the N determined by Kjeldhal's method
Dry residue of the alcoholic solution obtained from the neutral aqueous fraction
N determined by Kjeldhal's method on the aforesaid alcoholic residue
Sum of the N's determined on the ecgonine and anhydroecgonine fractions separated chromatographically
Ratio between the ecgonine and the anhydroecgonine nitrogens separated chromatographically
Ecgonine: computed from the figures given in columns 6 and 8
Anhydroecgonine: computed from the figures given in columns 6 and 8
For measuring the optical rotation and the absorption spectrum, use was made of products obtained and purified by the methods described earlier in this paper; the purity of the anhydroecgonine and the ecgonine was verified by circular paper chromatography, using the technique of W. Ciusa and A. Buccelli [ (22)] described hereunder.
Anhydroecgonine exhibits only one absorption band at 213 mμ with ε = 9.350, while ecgonine is absolutely transparent in the interval between 200 and 300 mμ (in the same conditions, cocaine exhibits two absorption bands, at 233 and 274 mμ respectively).
Determination of the quantity of ecgonine and anhydroecgonine in the various samples of residues from the processing of coca paste was carried out by determining the nitrogen separately, by Kjeldahl's method, in the two stains yielded by chromatographic separation, and comparing the result with the total nitrogen determined in the sample considered.
Preparation of ecgonine
4 g cocaine hydrochloride was dissolved in 40 ml HC1 0.2 N and the solution reflux-boiled for about 120 hours. The cooled acid solution, which contains benzoic acid in suspension, was extracted with ethyl ether, then akalized, again extracted with ethyl ether (the ethereal solutions being discarded), neutralized to pH 6.0-6.5, and finally evaporated to dryness in a water bath.
The residue was repeatedly extracted with anhydrous ethyl alcohol and the alcoholic extract was evaporatedto dryness. The resultant residue, after two crystallizations from anhydrous alcohol, yielded 0.44 g desiccated ecgonine, the elemental analysis of which (findings in per cent: C: 52.50; H: 8.56; N: 6.81; for C 9H 15O 3N.H 20 calc.: in per cent: C: 53.19; H: 8.43; N: 6.89) showed the composition of monohydrate (molecular weight 203.2), despite drying at 120 °C for two hours over calcium chloride at a pressure of about 1 mm mercury.
Preparation of anhydroecgonine
This was carried out by the method described by A. Einhorn (2), the ecgonine being treated with POCl 3. The residue of anhydroecgonine iodohydrate, obtained after distillation in a stream of iodine vapour carried out on the solution of iodine iodohydrate and subsequent evaporation, crystallized twice from anhydrous ethyl alcohol, still exhibited a faint yellow coloration. After passage through an Amberlite I.R. 45 column, 1.90 g iodohydrate yielded 1.05 g anhydroecgonine base, which was crystallized once from anhydrous ethyl alcohol (findings in per cent: C: 64.35; H: 7.91; N: 8.35; for C 9H 13O 2N calc. in per cent: C: 64.65; H: 7.84; N: 8.38).
Paper chromatography of mixtures of ecgonine and anhydroecgonine
The technique described by W. Ciusa and A. Buccelli (22) was used.
At a distance of 2.5 cm from the centre of the paper disc, about 5-20 μl of a 1-2% solution of ecgonine and anhydroecgonine or of a mixture of the two in 95°ethyl alcohol is deposited.
The eluent is a mixture of isopropyl alcohol, water and sodium acetate, obtained as follows: 1.0 g sodium acetate is made up with water to 10 ml, and this solution is diluted to 50 ml with isopropyl alcohol.
The procedure, using only one phase, was such that during chromatography the solution divided into a faster hydroisopropanol solution (I) and a slower solution (II) which contained sodium acetate.
The 50 ml solvent so prepared is sufficient for four chromatographs. Using watch glasses 30 cm in diameter, and care being taken to avoid losses of solvent through evaporation, it proved possible to obtain good chromatographs after 12-15 hours without the leading face of the solvent reaching the edges. To achieve better separation of the two substances it is possible to allow the eluent to reach and flow over the edges of the glasses by prolonging the operation for 8-10 further hours.
After drying, the paper was exposed to the light from a Philips germicidal lamp with a Corning 9863 glass filter; this made it possible to observe the anhydroecgonine stains alone, just in front of the leading face of solution II; exposure to iodine vapour for 5-10 minutes brings out not only the anhydroecgonine stains, but the ecgonine stains as well, with a slightly lower R F. To achieve stable coloration of the two stains, after the iodine has been allowed to sublimate, the paper is sprayed with a solution of Dragendorff's reagent diluted in the proportion of 1:4 with acetic acid. The anhydroecgonine stain then takes on a brownish-pink and the ecgonine stain a pale pink coloration.
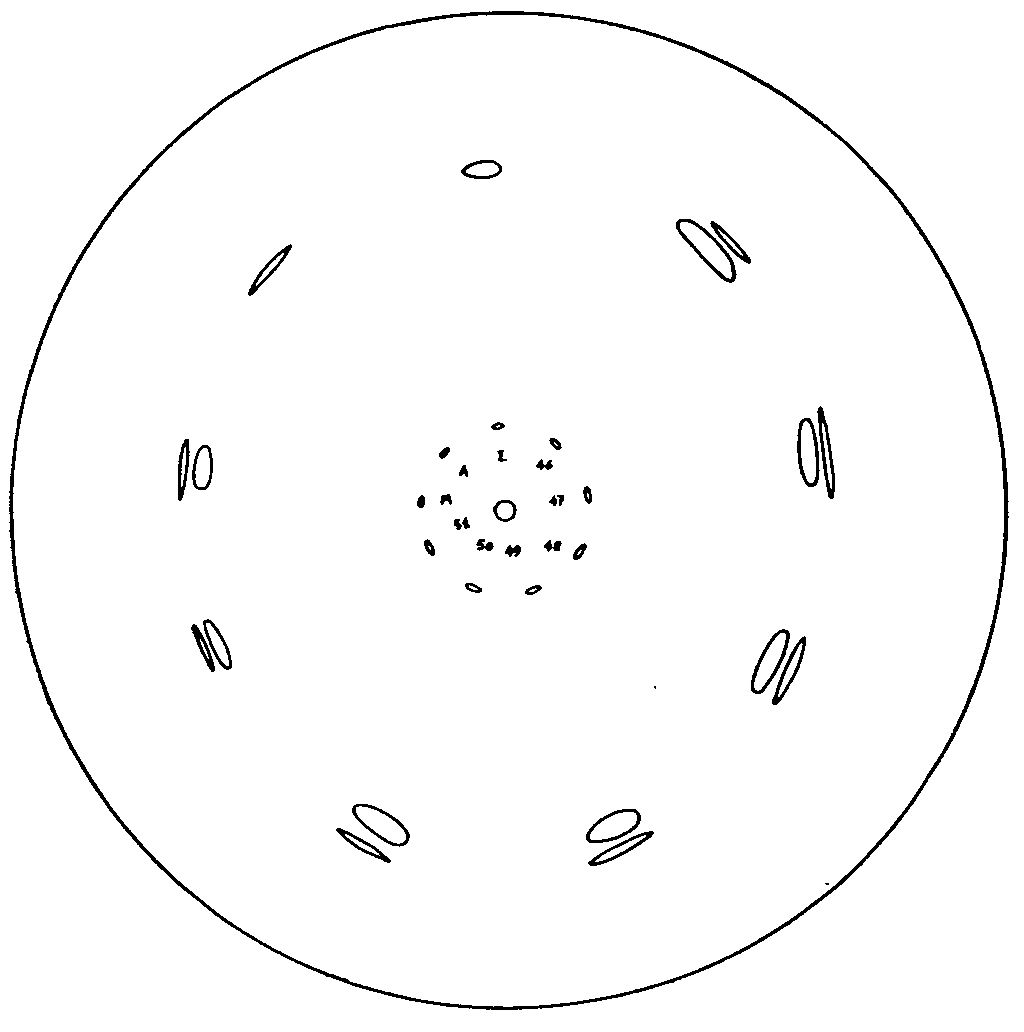
The R F's calculated in the described conditions for an ambient temperature between 18 °C and 22 °C were respectively as follows: 0.37 for ecgonine, and 0.40 for anhydroecgonine (the advancing face of solution II, containing sodium acetate, being at 0.41); although close together, the two stains are clearly separated.
Quantitative determination was performed by cutting the paper around the stains and determining, by Kjeldhal's method, the total nitrogen in the cuttings. The figures obtained were corrected for the nitrogenous impurities of the base as determined on the paper surrounding the stains (about 0.34-0.36 mg nitrogen per g of paper).
Figure 1 shows a typical chromatogram obtained with ecgonine, anhydroecgonine, and six different samples of residues from the preparation of cocaine from coca paste.
Ultraviolet spectra of ecgonine and anhydroecgonine
The absorption spectra of cocaine, ecgonine and anhydroecgonine between 210 and 300 mμ in decinormal HC1 were determined. The cocaine spectrum corresponds closely to that reported in the literature (16). Below are given the maximum and minimum molecular absorption values ε:
Ecgonine: no absorption between 210 and 300 mμ
Anhydroecgonine: maximum at 213 mμ: 9.350;
Cocaine: maxima: at 233 mμ: 13.000; at 274 mμ:
130; minimum: at 262 mμ: 860
A very slight absorption noted between 210 and 220 mμ in our sample of ecgonine is manifestly due to the presence of an anhydroecgonine impurity in the proportion of about 0.5 per cent; pure ecgonine exhibits no absorption between 210 and 300 mμ.
The absorption spectra were obtained with a Beckman DU spectrophotometer in 1000-cm 3 flasks with a slit of 0.3-0.6 mm. The ε values are referred to the base dissolved in decinormal HCl.
Polarimetric measurements of solutions of ecgonine and anhydroecgonine at various pH-values and deduction of the constants of acidic and basic dissociation
Table 3 shows the values of the optical rotations computed from our polarimetric measurements carried out at 27-29°with a sodium lamp in a 10-cm capillary tube.
The ecgonine and the anhydroecgonine were prepared and purified by the methods described.
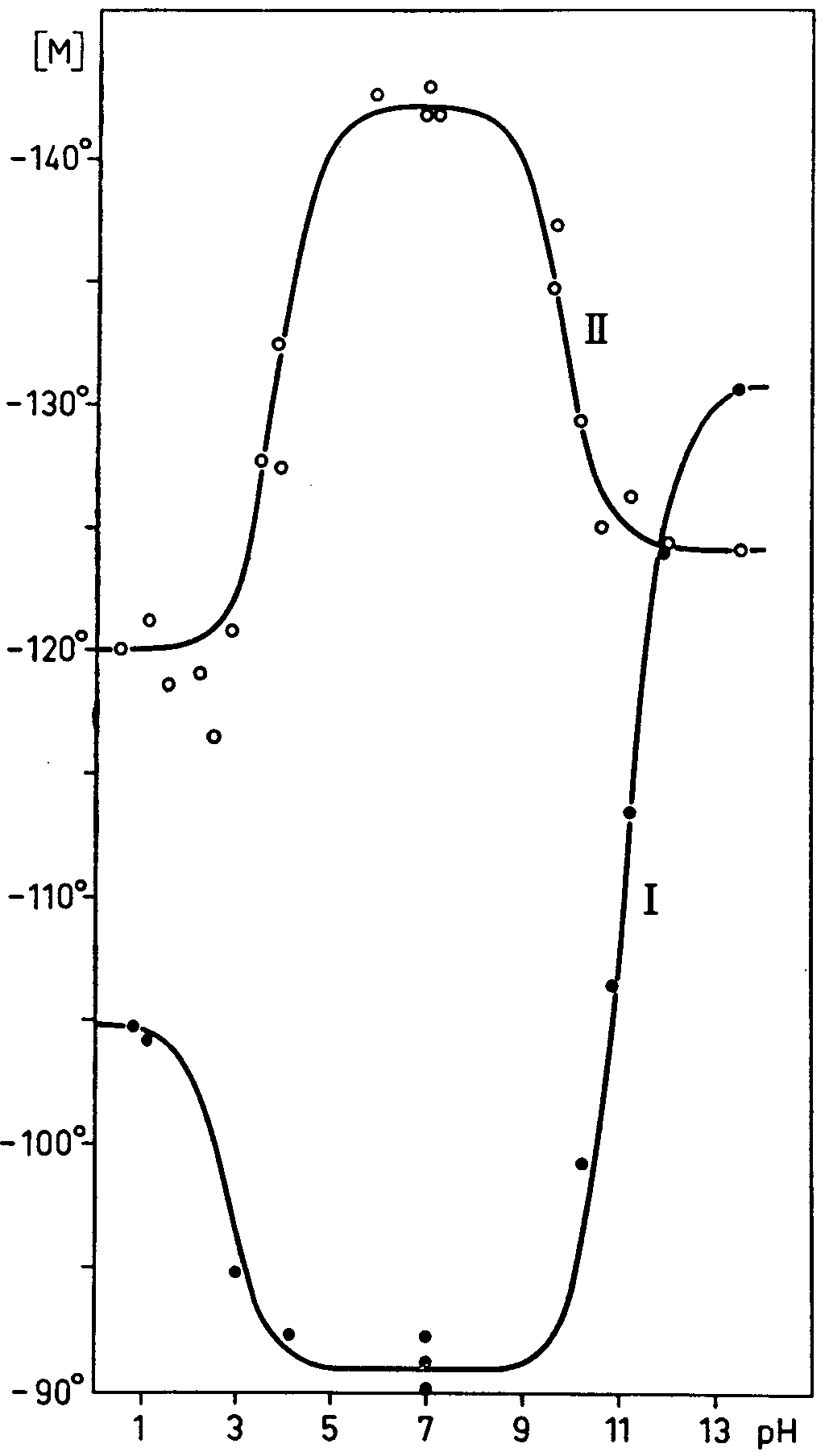
The two diagrams in figure 2 show the computed course of the optical rotation (see table 3) which more closely approaches the experimental course.
Coca paste. Residues from the industrial extraction of coca ine. Ecgonine and anhydroecgonine 33
|
Substance |
Molecular weight |
[α]D |
[M]D |
Reaction |
|---|---|---|---|---|
|
Ecgonine
|
203.2 |
-44.8°
|
-91.0°
|
Neutral
|
|
Ecgonine
|
203.2 |
-51.6°
|
-104.8°
|
Strongly acid
|
|
Ecgonine
|
203.2 |
-64.2
|
-130.6°
|
Strongly alkaline
|
|
Anhydroecgonine
|
167.2 |
-85.0°
|
-142.1o
|
Neutral
|
|
Anhydroecgonine
|
167.2 |
-71.8°
|
-120.0°
|
Strongly acid
|
|
Anhydroecgonine
|
167.2 |
-74.3°
|
-124.2°
|
Strongly alkaline
|
For the anhydroecgonine no racemizing action due to the alkaline medium was observed; after a slight reduction in the optical rotation observed after one hour ([α] D= -73.0°; [M[D = -122.0°), no further variation was observed in the course of fifteen days at ambient temperature.
From our measurements carried out with the Beckman G potentiometer verified with solutions of known pH for the acidic, neutral and basic medium, the following can be quoted: for ecgonine pK a =11.1 and pK b = 11.2, in complete conformity with the results obtained by Kolthoff by the colorimetric method (10); for anhydroecgonine pK a = 9.8 (Kolthoff gives 10.1) and pK b = 10.2 (Kolthoff gives 10.85 and Veley (6) 10.42). It is noteworthy that the presence in the anhydroecgonine of the double link associated with the carboxyl (and perhaps also the consequent change in form of the molecule) substantially increased (by about one unit of pK) the apparent acidic force and the apparent basic force with respect of the ecgonine.
It is possible that the higher value of the true dissociation constants of the acidic and basic functions, and therefore the greater polar character of the molecule, should be attributed to the capacity possessed by ecgonine, but not by anhydroecgonine, tenaciously to hold on to a molecule of water.
Polarimetric measurement of liquids yielded by the acid hydrolysis of cocaine
Hydrolysis of cocaine hydrochloride yielded, depending of the concentration of the acid and the duration of boiling, the results set out in table 4, respectively expressed as: ( a) specific rotatory power referred to ecgonine hydrochloride (molecular weight 221.7); ( b) specific rotatory power referred to ecgonine (anhydrous, molecular weight 185.2); and ( c) molecular rotatory power.
As stated, our results confirm those reached by Nicholls (13).
|
[α]D |
|||
|---|---|---|---|
|
Conditions of hydrolysis |
Ecgonine hydrochoride |
Ecgonine |
[M] D |
|
1 hour, HC1 2N
|
-64.8°
|
-77.6°
|
-143.6°
|
|
5 hours, HCl 2N
|
-47.30
|
-57.0°
|
-105.7°
|
|
10 hours, HC1 2N
|
-47.9°
|
-57.3°
|
-106.2°
|
|
1 hour, HCl 6N
|
-50.9°
|
-60.9°
|
-112.8°
|
Comparison of the optical rotation in a strongly acid medium ([α] D = -57.0°; [M] D = -105-7°) attributed to the ecgonine yielded by total hydrolysis of cocaine with boiling HCI 2 N with the value for the rotation of pure ecgonine in the same conditions ([α] = -56.5°; [M] D = -104-8°)would suggest that about 6 per cent anhydroecgonine is formed by such hydrolysis; however, this value, obtained by indirect calculation, is only a very rough approximation. The fact remains that, as has been shown by the chromatographic examinations described, anhydroecgonine is formed in this hydrolysis.
The proportion of anhydroecgonine which is formed in the hydrolysis of cocaine depends on the concentration of the acid; it is relatively high with highly concentrated hydrochloric acid, and minimal (traces only, not discernible by chromatography, but only by spectrography) by boiling continued for over 100 hours with 0.2 N hydrochloric acid.
Lastly, since the values for the molecular optical rotation of ecgonine and anhydroecgonine respectively in a strongly acid medium do not differ greatly (-104.8°and -120.0°respectively), the approximate determination (error less than ± 7 per cent) of ecgoninic alkaloids by measuring the optical rotation in liquids of acid hydrolysis or in the hydrolized residues from the processing of coca leaves or coca paste is admissible. To achieve a more accurate result it would be necessary to know (whether specifically or not the relationship between anhydroecgonine and ecgonine, and accordingly to use a suitable coefficient included between the following extreme values for ecgonine and anhydroecgonine: [α] D= -56-5°([M] D = -104.8°) and [α] D= -64.8° ([M] D = -120.0°)·
The values of -56.5°and -64.8°were obtained from the corresponding values in table 3 (-51.6°and -71.8°respectively) or from the molecular rotations, and are both referred to non-hydrated ecgonine (molecular weight 185.2), while the aforesaid original values are referred as follows: -51.6°to ecgonine monohydride (molecular weight 203.2) and -71.8°to anhydroecgonine (molecular weight 167.2).
A similar commentary can be repeated for the readings on strongly alkaline solutions; for solutions recently alkalized the following values, calculated as above, are found for ecgonine and anhydroecgonine respectively: [α] D= -70.5°([M] D = -130.6°) and [α]D = -67-1°([M] D = -124.2°).
With the same solutions, during the first hour after alkalization, at ambient temperature, the above values are slightly lowered in the case of anhydroecgonine: [α] D = -66.0°; [M] D = -122.0° In these conditions, using the mean values ([M] D= - 127-40and - 126.3°respectively), the results computed from the polarimetric determinations should be correct to within about ± 3 %.
About 3 g of sample, accurately weighed, was dissolved in about 30 ml of water acidified with hydrochloric acid; the turbid solution was washed twice with an equal volume of ethyl ether, which was discarded: the aqueous phase was alkalized with NaOH and extracted twice with 45 and 20 ml respectively of ethyl ether. These last ethereal solutions, washed, mixed together and dried on sodium sulfate, yielded the residues indicated opposite No. 3 in table 2 (expressed in g per 100 g of undried sample).
Only on the more abundant basic residues, that is to say, on those obtained from the three samples Nos. 46, 47 and 51, were the determinations of total nitrogen by the Kjeldahl method and the chromatographic examination by the method described above (the details are omitted) were carried out; the latter revealed the almost complete absence of cocaine and the presence of a component having the spectrographic properties of methyl anhydroecgonine.
For the aforesaid samples Nos. 46, 47 and 51, the concentrations expressed in grammes of methyl anhydroecgonine computed from the aforesaid determination of nitrogen in the basic residue per 100 g of original sample are: 1.09, 3.67 and 3.92.
The aqueous fraction remaining after extraction with ether was neutralized to pH 6.0-6.5, evaporated to dryness in a water bath, and repeatedly extracted, with anhydrous ethyl alcohol. On aliquot portions of these alcoholic extracts, mixed together and made up to a volume of 50 ml, the dry residue was obtained and the chromatographic separations were performed.
|
Rotation (1g/1 ml) |
Experimental |
Computed |
|||||
|---|---|---|---|---|---|---|---|
|
No. of sample |
Total ecgonin in % |
acid |
neutral |
basic |
acid |
neutral |
basic |
| 46 | 12.75 |
-11.1°
|
-10.6°
|
-11.8°
|
-7.98°
|
-8.78°
|
-8.70°
|
| 47 | 32.0 |
-22.8°
|
-28.5°
|
-25.0°
|
-20.2°
|
-22.8°
|
-21.7°
|
| 48 | 28.0 |
-18.2°
|
-17.65°
|
-17.44°
|
-17.6°
|
-19.7°
|
-19.05°
|
| 49 | 25.1 |
-25.2°
|
-28.6°
|
-24.7°
|
-15.9°
|
-18.8°
|
-17.05°
|
| 50 | 9.0 |
-7.72°
|
-7.76°
|
-8.77°
|
-5.66°
|
-5.91°
|
-6.39°
|
| 51 | 10.3 |
-7.60°
|
-7.84°
|
-8.20°
|
-6.35°
|
-6.80°
|
-7.06°
|
The presence of ecgonine and anhydroecgonine was recognized and the quantitative determinations of them were carried out through the determinations of nitrogen on the stains yielded by chromatographic separation. The results are set out in table 2, under Nos. 7 to 10.
It should be observed that, within the methods' limits of error, the whole of the total alcohol of the water-soluble and alcohol-soluble residue, after alkaline extraction with ether, is found as the sum of the nitrogens of the ecgonine and the anhydroecgonine.
In order to ascertain whether the results obtained by us in the optical rotation of ecgonine and anhydroecgonine can be applied to the polarimetric determination of the total ecgonine in the residues from the preparation of cocaine from coca paste, the following procedure was applied.
About 2 g of residue (solid, semi-solid or liquid), accurately weighed, is dissolved in about 40 ml of water, the solution is acidified if necessary, and extraction is carried out three times with 20, 15 and 15 ml respectively of ethyl ether, which is discarded; after alkalization, extraction is performed as above with ethyl ether (basic fraction), followed by neutralization to pH 6.5-7.5, evaporation in the water bath, and drying of the residue; the latter is repeatedly extracted with a total of 40 ml of hot anhydrous ethyl alcohol, which is filtered and evaporated to dryness; the residue is taken up with water and made up to 10 ml. A polarimetric reading is taken; after strong acidification (pH<1.0) a further polarimetric reading is taken (in the computation account is taken of the increase in volume); after strong alkalization (pH>13.0) a further polarimetric reading is taken (the correction being performed as above).
In table 5, the aforesaid rotations are all referred to the concentration of 1 g of original residue examined per 1 ml.
The same table also shows the rotations and the total ecgonine (the sum of the ecgonine and the anhydroecgonine expressed as ecgonine anhydride of molecular weight 185.2), calculated from the composition of the residues as shown by the figures in columns 6 and 8 of table 2 and from the specific (or molecular) rotations shown in table 3: the rotations ascertained are almost all higher than those computed; more specifically, they range from 10% below to 60% above (the mean being 20% above).
It is concluded from the foregoing that polarimetric measurements do not enable the total ecgonine of these residues to be determined; at all pH values, these residues behave as though they contained variable quantities of a substance more strongly levorotatory than either ecgonine or anhydroecgonine.
Instituto Superiore di Sanité (Higher Institute of Health), Biological Laboratories. December 1964
Liebermann, C.: Ber. 21: 11,2342 (1888)
002Einhorn, A.: Ber. 22: 1495 (1889)
003Hesse: Pharm. J. (3) 21: 1112 (reported by Beilstein, 22: 196)
004Greshof, M.: Pharm. Weekbl., 44: 961 (1907); C. 11: 1023 (1907)
005De Jong, A. W. K.: Pharm. Weekbl., 45: 42 (1908); C. 1908: I, 559 (1908)
006Veley, V. H.: J. Chem. Soc., 93: 652 (1908)
Carr, F. H., and Reynolds, W. C.: J. Chem. Soc., 97: 1328 (1910)
008Bierling, E., Pape, K., and Viehover, A.: Arch. Pharm., 248: 303 and 336 (1910)
009De Jong, A. W, K.: Rec. Trav. Chem., P.B., 42: 980 (1923)
010Kolthoff, J. M.: Biochem. Z., 162: 289, 311, 312 (1925)
011Chemnitius, F.: J. Prakt. Chem., 116: 285 (1927)
012Castille, M. A., and Ruppol, E.: Bull. Soc. Chim. Biol., 10: 623, 635 (1928)
013Nicholls, J. R.: Analyst, 61:155 (1936)
014Nicholls, J. R.: Bulletin of the Health Organisation (League of Nations) 7: (3) 461 et seq. (1938)
015Gastaldi, E.: Boll. Chim. Farm., 88:175 (1949)
016Farmilo, C. G.: Bulletin on Narcotics, 6: 3-4, 20 (1954)
017Garrat, D. C.: The Quantitative Analysis of Drugs, second edition, London 1955, p. 140
018Leiserson, L. and Walker, T. B.: Analytical Chemistry, 27: 1129 (1955)
019White, E. H. and Dunathan: J. Am. Chem. Soc., 78: 6055 (1956)
020Asahina, H., and Ono, M.: Eisei Shikejo Hokoku, 75:113 (1957); C.A. 52:17613 (1958)
021Heller, W.: "Polarimetry" in Weissberger, A.: Physical Methods of Organic Chemistry, third edition, vol. 1: part III, New York 1960, p. 2300
022Ciusa, W., and Buccelli, A.: Rassegna Chimica, XII, 5 (1960)
023Bell, M. R., and Archer, S.: J. Am. Chem. Soc., 82: 4642 (1960)


