Experimental
Summary
Author: Elna NIEMINEN
Pages: 23 to 28
Creation Date: 1971/01/01
NOTE BY THE EDITOR: This paper, though it does not deal exclusively with narcotics or psychotropic substances, is of interest in presenting new analytical methods which could be applied by control laboratories for the identification and quantitative analysis of multi-component preparations.
The problem of devising an accurate method for assaying commonly used analgesic and antipyretic compounds in pharmaceutical formulations has become classical in the literature of pharmaceutical analysis. The methods employed have been reviewed by Haefelfinger et al ( [ 1] ), by Dibbern and Scholz ( [ 2] ) and by Clayton and Thiers ( [ 1] ). Most of the solutions proposed involve multiple steps, including lengthy and tedious liquid-liquid extractions, ion-exchange chromatography and/or column chromatography separations followed by quantitation of the separated ingredients by UV spectrophotometric, titrimetric or colorimetric methods. Direct spectrophotometric determinations have also been described ( [ 1] ) but the excipients employed in analgesic tablets may interfere with the quantitation.
In recent years gas-liquid chromatography (GLC) has proved an exceptionally valuable tool for simultaneous separation and quantitation of several constituents from a multicomponent mixture. Although numerous applications of this technique in the field of pharmaceutical analysis have already been described ( [ 1] ), its use for the determination of analgesic and antipyretic compounds in dosage forms has so far been very limited.
Hoffman and Mitchell ( [ 1] ) described a GLC procedure on a 2 per cent DC-200 column for determining acetylsalicylic acid, phenacetin, and caffeine in tablets. For the separation of caffeine and phenacetin combined with other active ingredients in tablets, a 10 per cent SE-30 column has also been used ( [ 1] ). With both methods it is difficult to obtain reproducible results, owing to the high ratio of phenacetin to caffeine and the small difference in their retention times. Dechene et al. ( [ 6] ) improved Hoffman's procedure and determined acetylsalicylic acid and phenacetin on a DC-200 column, and codeine and caffeine on a 10 per cent SE-30 column after liquid-liquid extraction. Other columns used for separating antipyretics are 1.6 per cent BDS and 0.8 per cent BDS + 1 per cent XE-60 columns ( [ 1] ). However, BDS has a maximum temperature limit of 180 °C and XE-60 of 250 °C.
This article is based on a paper which was presented at the meeting of the Commission of Control Laboratories, 13-14 April 1970, in Montpellier.
To improve the stability of the SE columns, new stationary phases have been produced by increasing the average molecular weights of the SE-30, SE-33, and SE-52 polysiloxanes. These are denoted as OV-1, UCC-W-982 and OV-17 and they have a maximum temperature limit of 300 °C. Recently, Alber ( [ 1] ) described a 3 per cent OV-17 column for use as a universal column for pharmaceuticals and reported retention times for salicylamide, phenacetin and caffeine.
We have successfully employed a 10 per cent UCC-W-982 column for the separation of several pharmaceuticals. The present paper describes the procedure we have used for the determination of components in analgesic tablets and dosage powders.
Instrumental conditions
The gas chromatograph used was a Hewlett-Packard Model 5750 B, equipped with a dual flame ionization detector and a Moseley Model H 10-7128A strip chart recorder with a disc integrator. The steel column 6' X 1/8" was packed with 10 per cent UCC-W-982 on 80-100 mesh Chromosorb W AW-DMCS and heat-conditioned at 250 ° for 48 hours and at 270 ° for three hours. Carrier gas : nitrogen, flow rate 30 ml/min. Detector gases : air, flow rate 300 ml/min and hydrogen, flow rate 30 ml/min .
Reference standards and reagents
Throughout this work Ph. Nordica substances were used as reference standards. All reagents were of analytical grade.
Methods for quantitative determinations
The procedure described below for tablets containing propylphenazone 150 mg, ethenzamide 150 mg, caffeine 50 mg, and codeine phosphate 7 mg, was used for all the samples. The concentrations of sample solutions, the internal standards, and the chromatographic parameters are given in the legends to the figures.
Assay of propylphenazone, ethenzamide, and caffeine
Internal standard - A 2 mg/ml solution of phenacetin in chloroform-methanol 1:3.
Mixed standard - Accurately weighed amounts of about 150 mg of propylphenazone, 150 mg of ethenzamide and 50 mg of caffeine were dissolved in 25 ml of internal standard.
Preparations of sample - An aliquot of the powder from the finely ground tablets equivalent to the average tablet weight was transferred to a 25-ml volumetric flask, 15 ml of internal standard was added, and the flask was immersed in an ultrasonic cleaning bath (Kerry, Ultrasonic Ltd) for 1-2 min. or shaken vigorously for 30 min. The flask was filled to the mark with internal standard, shaken and left to settle. The clear supernatant solution was used for chromatography.
Assay of codeine phosphate
Internal standard - A 2 mg/ml solution of histapyrrolidine hydrochloride in chloroform.
Standard solution - An accurately weighed amount of about 10 mg of anhydrous codeine dissolved in 2 ml of internal standard.
Sample preparations - An accurately weighed quantity of the powdered tablet equivalent to about 10 mg of codeine was transferred to a 100-ml beaker and mixed with 2 ml of water and 1 ml of 10 per cent ammonia to a uniform slurry. 3 g of Celite 545 was added and the mixture was ground until it was homogeneous and transferred quantitatively to a glass column, 19 mm X 350 mm. Elution was carried out with 125 ml of chloroform and the eluate was collected in an Erlenmeyer flask containing 20 ml of 0.1 N HCI. The chloroform layer was separated, washed three times with 10 ml of 0.1 N HCI and discarded. The combined aqueous layers were made alkaline with 10 per cent ammonia and extracted four times with 25 ml of chloroform. The chloroform layers were filtered through anhydrous Na2SO4 and evaporated to dryness. The residue was dissolved in 2 ml of internal standard.
Assay of single tablets
The procedure described above was used as follows : one tablet was transferred to a 50-ml glass-stoppered centrifuge tube containing an accurate volume of internal standard, and dispersed by immersing the tube for 1-2 minutes in an ultrasonic cleaning bath.
Results and discussion
The GLC of some commercially available tablets and a dosage powder are presented in figs. 1-7. The seperation was excellent for all the components except acetylsalicylic acid. On a UCC-W-982 column this gave only a small peak and the results were not reproducible. In addition, we found that if both acetylsalicylic acid and salicylamide were present in dosage forms, the acetylsalicylic acid interfered with the separation of the salicylamide.
Codeine phosphate must first be separated from the sample as the base, after which it can be chromatographed on the same column (fig. 6).
Because liquid-liquid extraction often leads to the formation of emulsions, our procedure in routine analysis has usually been first to pass the sample through an ammonia-impregnated Celite 545 column. After column chromatographic separation, liquid-liquid extraction has been carried out to remove most of the other active ingredients.
The most time-consuming step in the procedure is to discover an appropriate internal standard and to select the chromatographic settings so that all the components can be analysed simultaneously. The use of an internal standard, however, greatly simplifies the analytical procedure and calculations. In addition, it minimizes the error due to the variation in injection volume. It was found that when the injection volumes were 1 μl, 2 μl and 3 μl, the variation in the ratios of the sample peak area to the internal standard peak area was less than 2 per cent.
The results obtained in the quantitative determinations are presented in tables 1 and 2. The amounts found varied between 93 and 108 per cent of the labelled declarations and were thus within the accepted limits. The procedure is especially suitable for the control of content uniformity of tablets and dosage powders.
For three months we have used the same UCC-W-982 column daily and it has not shown any sign of deterioration. In addition to the analysis of ingredients in analgesic formulation, the column has been used for the determination of antihistamines, amphetamine, amitriptyline, and atropine. It seems to be a very good standard column for pharmaceuticals.
|
Sample |
Active components |
Labelled mg/tablet |
Found mg/tablet |
|---|---|---|---|
|
Tablet A
|
Propylphenazone
|
150 | 160 |
|
Ethenzamide
|
150 | 152 | |
|
Caffeine
|
50 | 52 | |
|
Codeine phosphate
|
7 | 6.7 | |
|
Tablet B
|
Propylphenazone
|
200 | 210 |
|
Salicylamide
|
200 | 195 | |
|
Caffeine
|
50 | 54 | |
|
Codeine
|
10 | 9.5 | |
|
Tablet C
|
Acetylsalicylic acid
|
200 |
205
a
|
|
Phenacetin
|
200 | 203 | |
|
Codeine phosphate
|
10 | 10 | |
|
Caffeine
|
50 | 52 | |
|
Phenobarbital
|
25 | 27 | |
|
Tablet D
|
Amidopyrine
|
300 | 319 |
|
Phenobarbitone
|
50 | 53 | |
|
Codeine phosphate
|
30 | 28 | |
|
Caffeine
|
100 | 105 | |
|
Tablet E
|
Salicylamide
|
200 | 189 |
|
Paracetamol
|
200 | 197 | |
|
Caffeine
|
50 | 48 | |
|
Codeine phosphate
|
10 | 9.3 | |
|
Phenobarbital
|
25 | 24 |
a Determined colorimetrically.
|
Phenazone |
Ethenzamide |
Caffeine |
|
|---|---|---|---|
|
Labelled mg/powder
|
700 | 200 | 100 |
|
Found mg/powder
|
|||
|
Mixture of:
|
|||
| 10 powders | 707 | 195 | 98 |
| 1 powder | 715 | 204 | 105 |
| 1 powder | 692 | 209 | 102 |
|
Mean
|
705 | 203 | 102 |
A gas-chromatographic method is described for simultaneous separation and quantitation of some commonly used antipyretics, phenobarbital, and caffeine in analgesic preparations. The determinations were carried out on a 10 per cent UCC-W-982 column using an internal standard technique. Codeine phosphate was first separated from the samples as the base, after which it was chromatographed on the same column. The results obtained varied between 93 and 108 per cent of the labelled amounts.
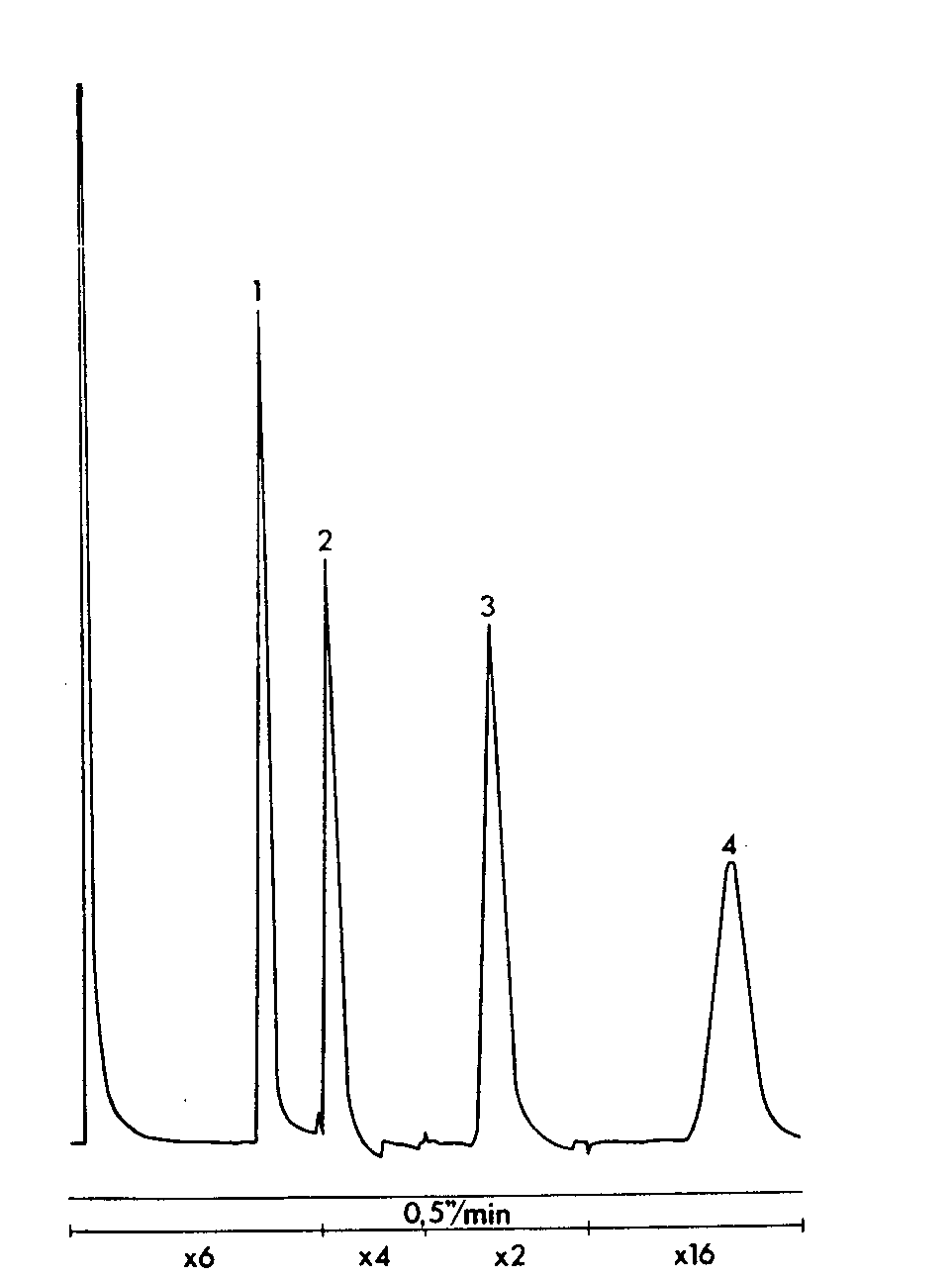
GLC separation of ethenzamide, caffeine and propylphenazone (Tablet A).
Sample: 1 tablet in 25 ml of internal standard, 2 μl injection volume. Column temperature 195 °C, injection port and detector temperatures 300 °C. Sensitivity range 10 3. Attenuation and chart speed below chromatogram.
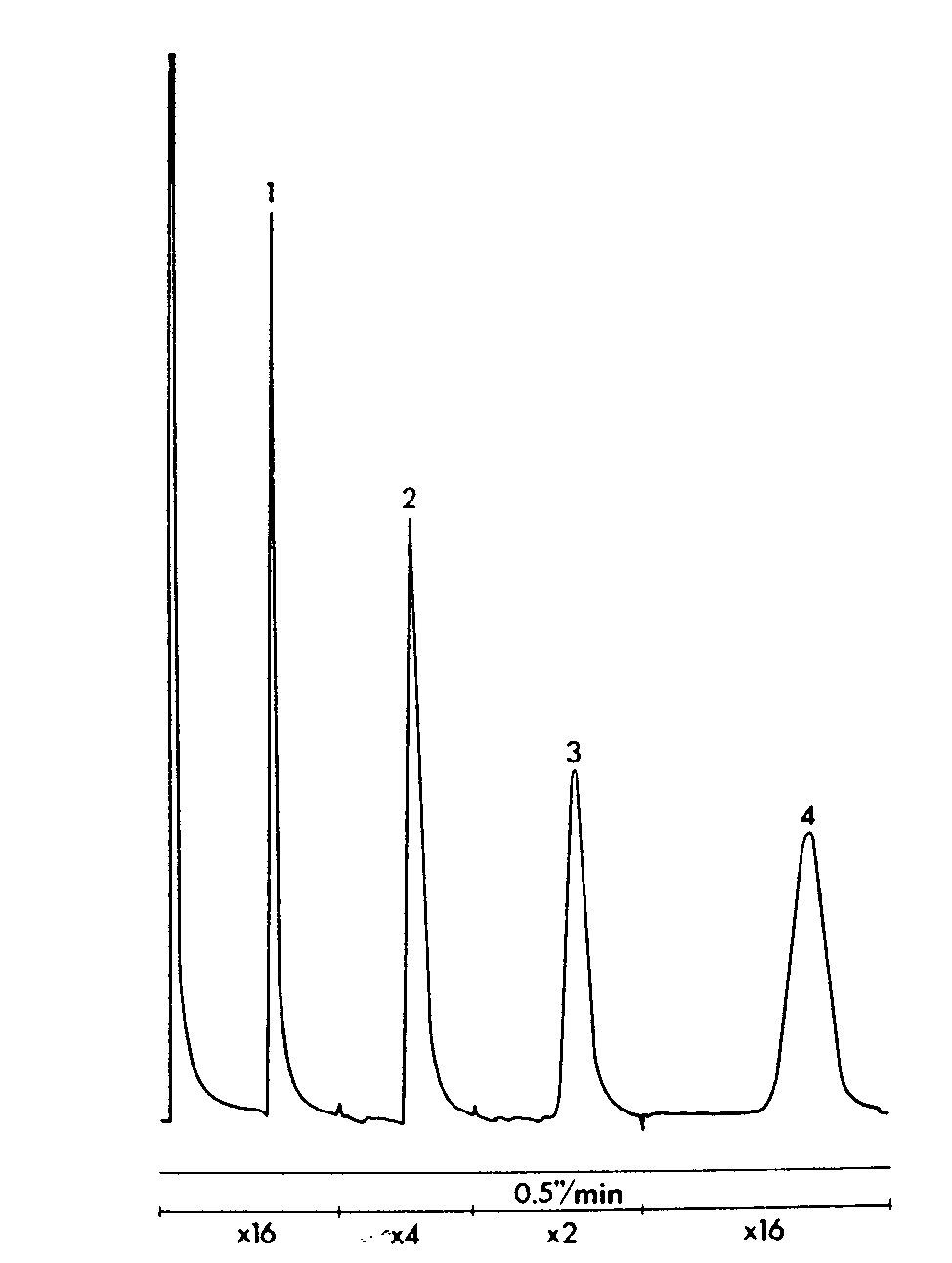
GLC separation of salicylamide, caffeine and propylphenazone (Tablet B)
Sample: 1 tablet in 25ml of internal standard,2μl injection volume. Column temperatures 195°C, injection port and detector temperatures 300 °C. Sensitivity range 10 3. Attenuation and chart speed below chromatogram.
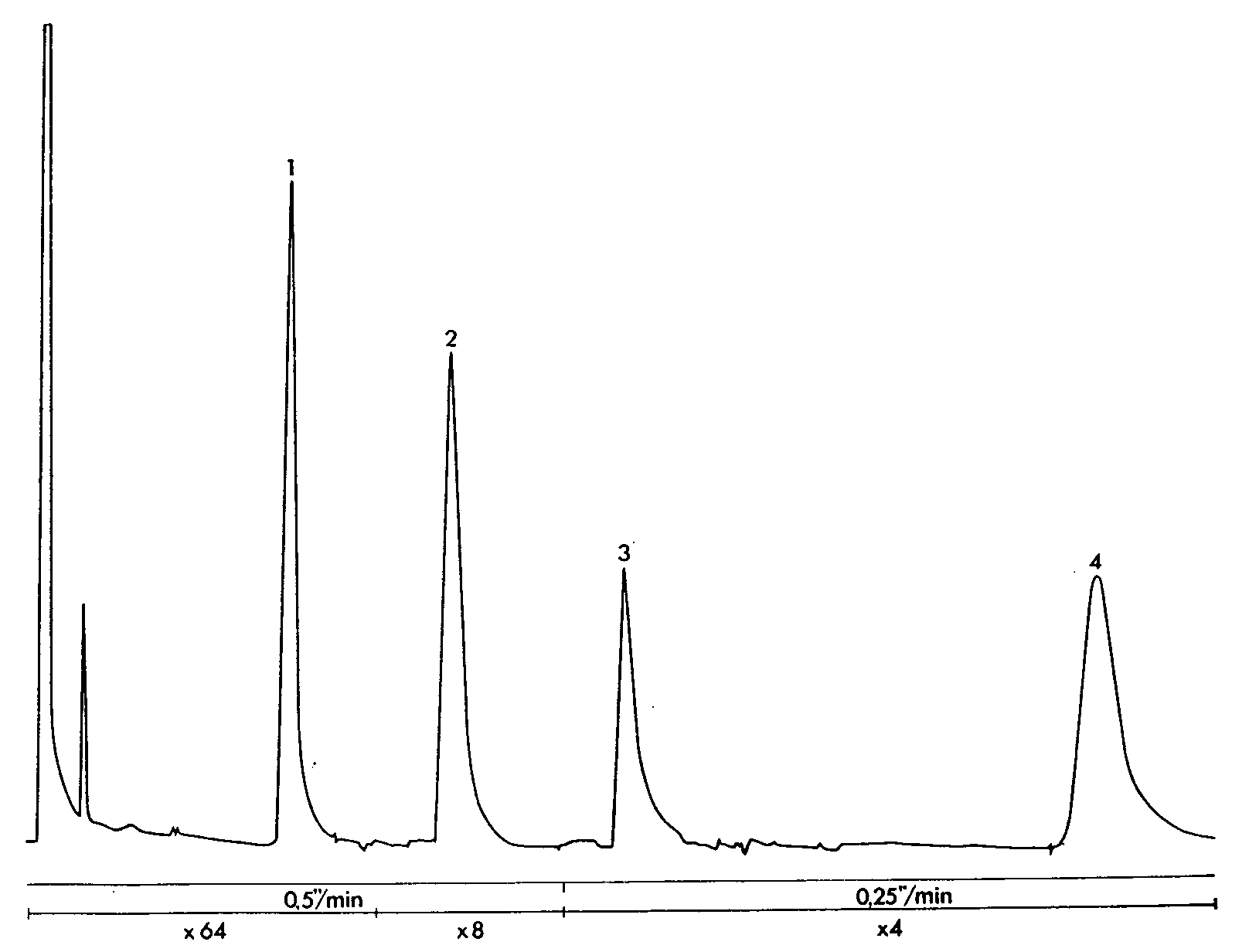
GLC separation of phenactin, caffeine and phenobarbital (Tablet C)
Sample: 1 tablet in 25 ml of internal standard, 2 μl injection volume. Column temperature 200°C, injection port and detector temperatures 300 °C. Sensitivity range 10 2, attenuation and chart speed below chromatogram.
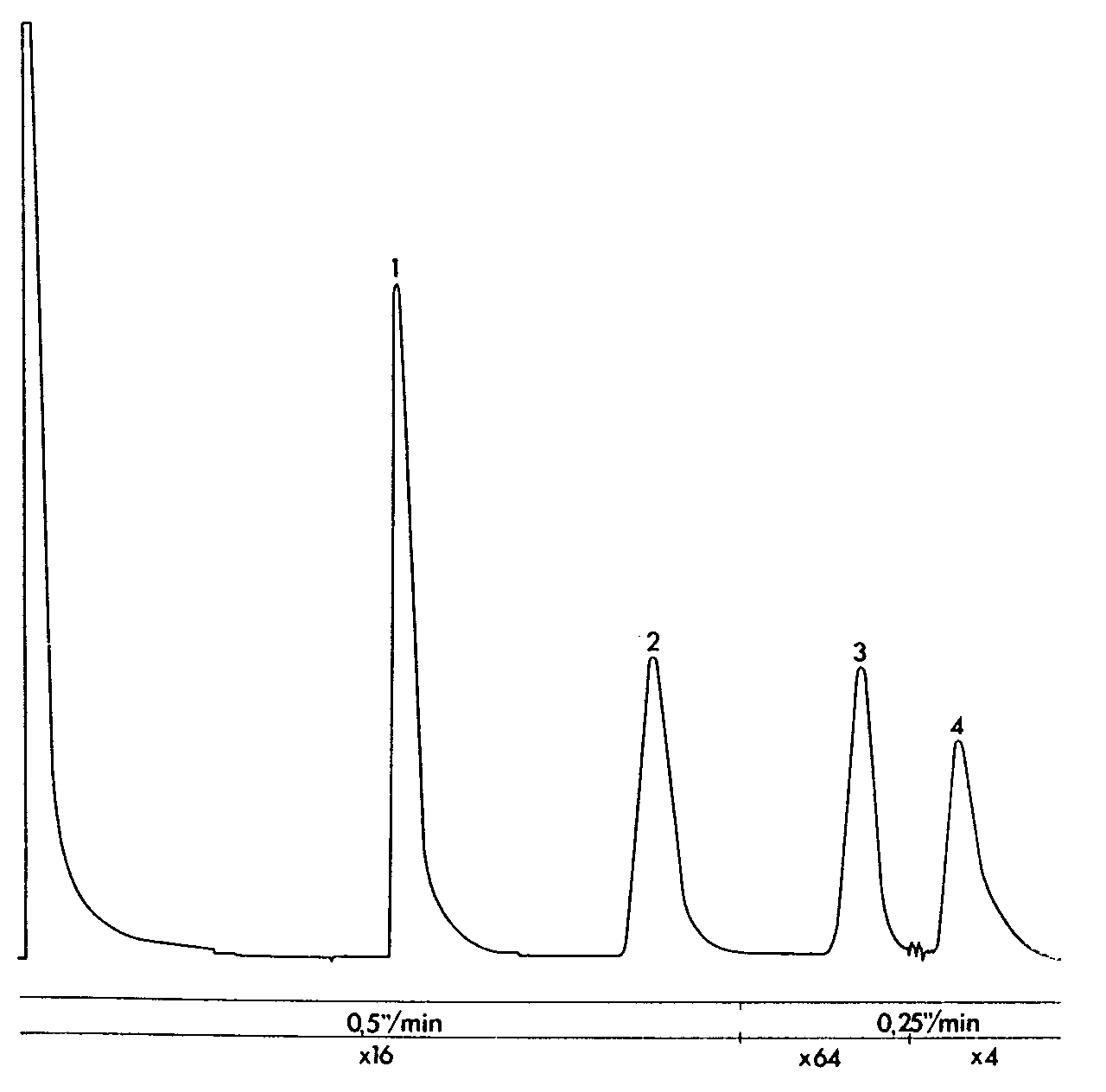
GLC separation of caffeine, aminophenazone and phenobarbital (Tablet D)
Sample: 1 tablet in 25 ml of internal standard, 2 μl injection volume. Column temperature 175 °C, injection port and detector temperatures 300 °C. Sensitivity range 10 2, attenuation and chart speed below chromatogram.
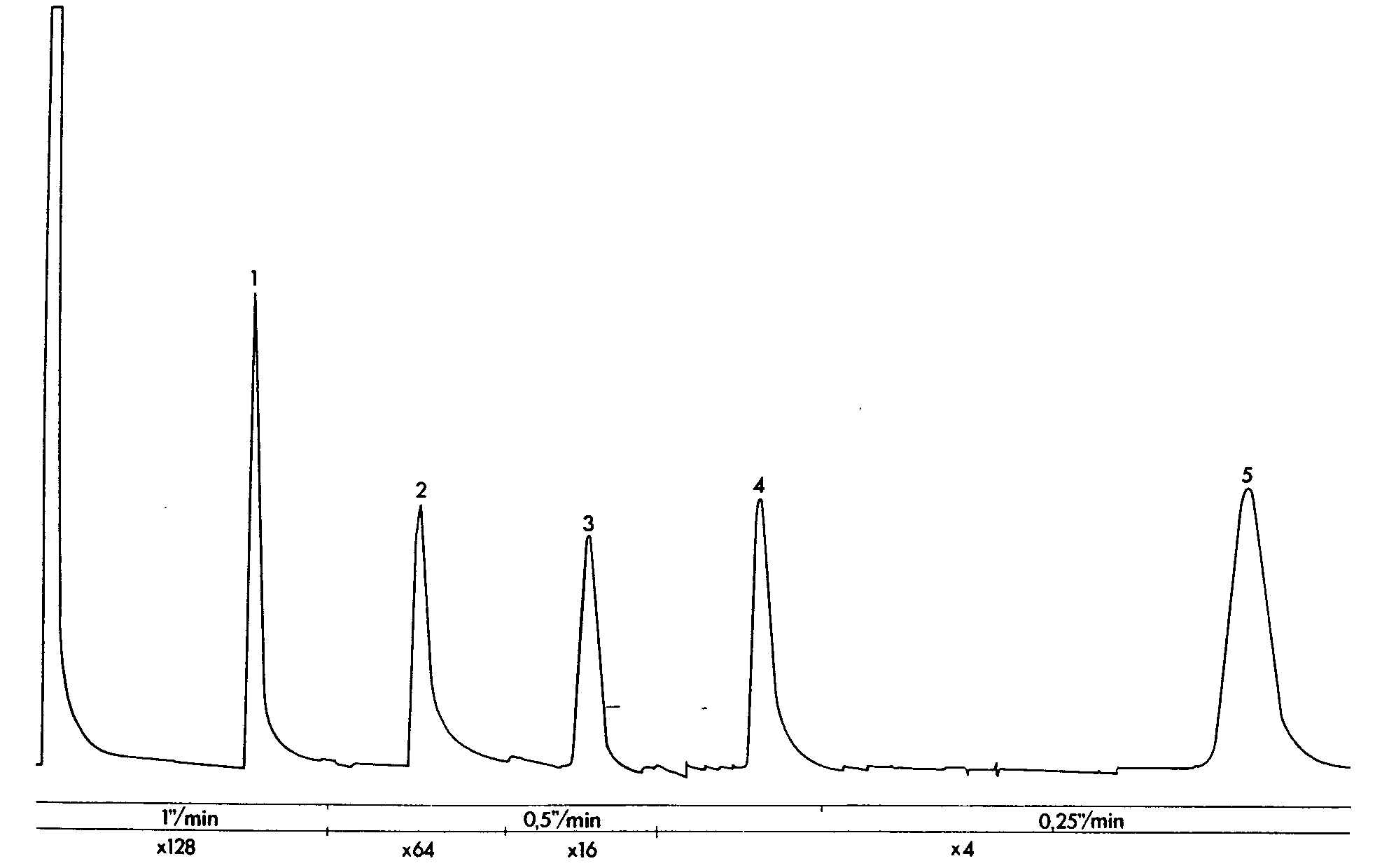
GLC separation of salicylamide, paracetamol, caffeine and phenobarbital (Tablet E)
Sample: 1 tablet in 25 ml of internal standard, 2 μl injection volume. Column temperature 195 °C, injection port and detector temperatures 300 °C. Sensitivity range 10 2, attenuation and chart speed below chromatogram.
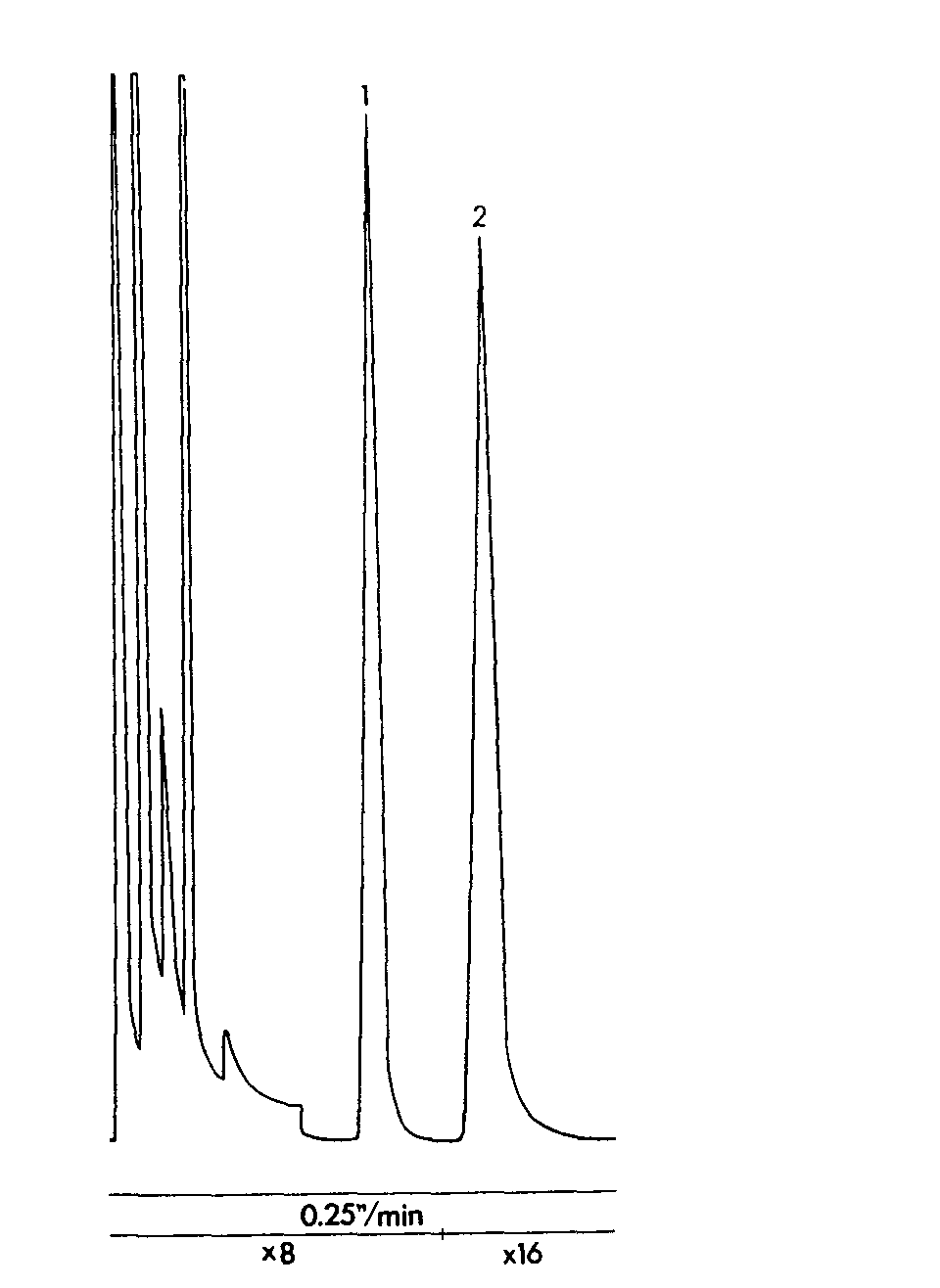
GLC of codeine separated from Tablet E
Sample: about 10 mg in 2 ml of internal standard, 2 μl injection volume. Column temperature 240 °C, injection port temperature 300 °C and detector temperature 270 °C. Sensitivity range 10 2. Attenuation and chart speed below chromatogram.
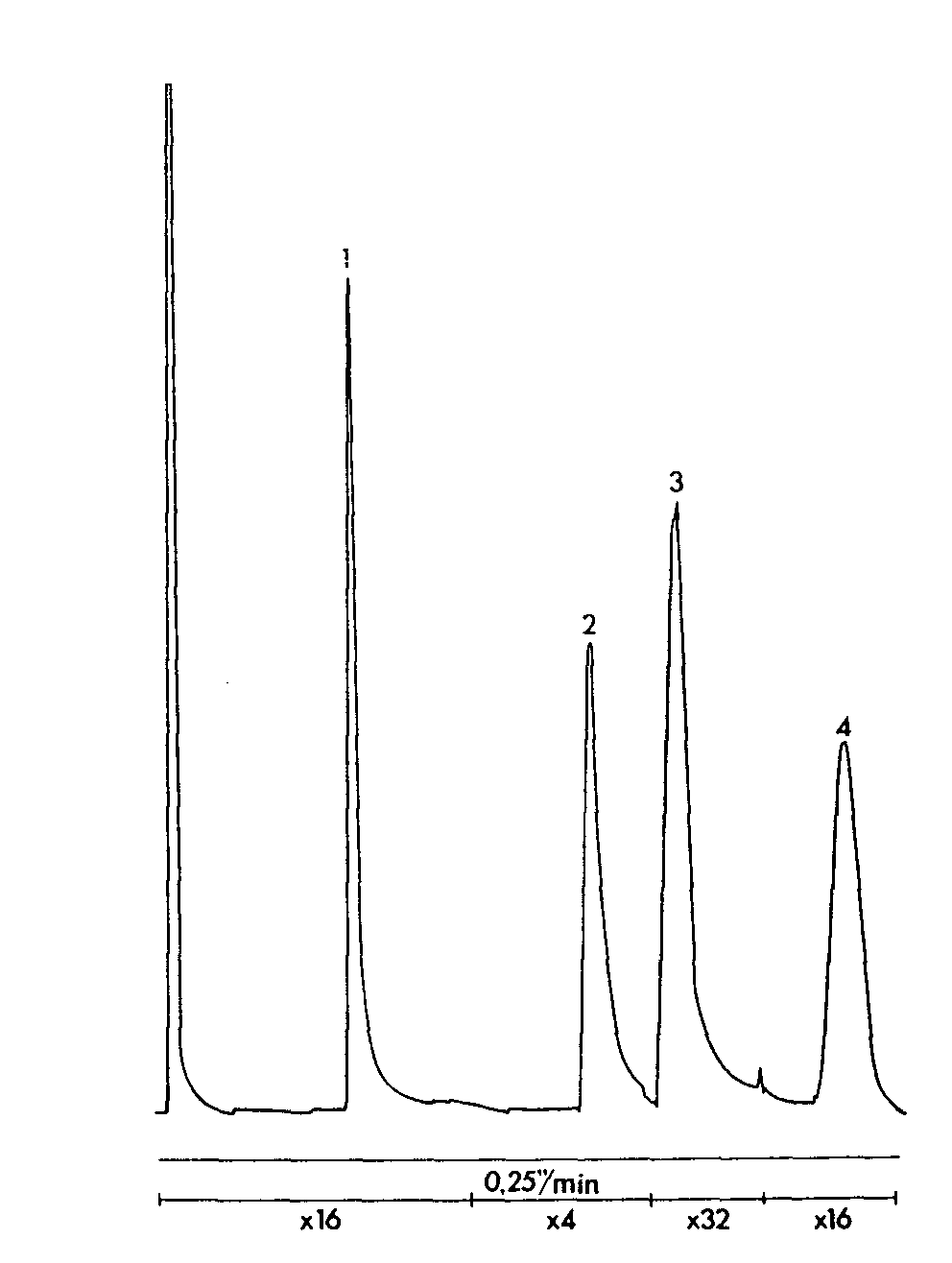
GLC separation of ethenzamide, caffeine and phenazone
Sample: 1 dosage powder in 50 ml of internal standard, 2 μl injection volume. Column temperature 185 °C, injection port and detector temperatures 300 °C. Sensitivity range 10 2. Attenuation and chart speed below chromatogram.
P. Haefelfinger, B. Schmidli and H. Ritter, Arch. Pharmaz. 297, 641-648 (1964).
002H. W. Dibbern, and G. Scholz, Arch. Pharmaz. 298, 175-184 (1965).
003A. W. Clayton and R. E. Thiers, J. Pharm. Sci. 55, 404-407 (1966).
004B. J. Gudzinowicz, Gas Chromatographic Analysis of Drugs and Pesticides, vol. 2. Marcel Dekker, Inc., N.Y., 1967.
005A. J. Hof
006fman and H. I. Mitchell, J. Pharm. Sci, 52, 305-306 (1963)
007A. Monard, J. Pharm. Belg., 23, 323-332 (1968)
008L. L. Alber, J. Assoc. Offic. Anal. Chemists. 52, 1295-1300 (1969).

