Introduction
Materials and methods
Extraction from urine
Silylation procedure
Gas chromatography
Quantitative determination of drugs excreted in rat urine
Results and discussion
Author: Yuji MARUYAMA, Aiko SAWA, Eikichi HOSOYA
Pages: 37 to 45
Creation Date: 1975/01/01
Thin-layer chromatography (TLC) is a very popular method for the detection of several narcotics and other dangerous drugs in human urine [ 1] [ 2] [ 3] [ 4] [ 5] [ 6] [ 7] [ 8] [ 9] [ 10] [ 11] . The main reasons for using TLC are its economy, its simplicity and its speed, which make it possible to handle many samples at the same time. However, it is not always a suitable method, especially for the identification of unknown compounds using only RF values. Other disadvantages are lack of specificity and sensitivity as well as the frequent interference of other substances present in blood and urine. A second screening by gas chromatography (GC) or GC-Mass spectrometry (GC-MS) is commonly required in order to prove the results. Procedures for the identification of drugs in biological samples by GC and GC-MS were developed by our laboratory and reported elsewhere [ 12] [ 13] [ 14] [ 15] [ 16] . In the present paper, methodologies are summarized for the determination of drugs such as methamphetamine (MP), pethidine (PE), morphine (M), nalorphine (N), codeine (CD), cocaine (CC), methadone (MT) and pentazocine (PZ) by simple extraction and purification for analysis by GC.
Chemicals. All chemicals were of reagent grade, obtained from Kanto-Kagaku or Wako Chemical Industries, Ltd.; the TMS-BA was procured from Tokyo Kasei Kogyo Co., Ltd. The drugs used were bought legally from a pharmaceutical company in Japan.
Animals. Male Wistar rats (Nihon-Rat Co.) weighing 100-150 g were used in the experiments.
Twenty millilitres (μl) of urine was poured into a column (1 cm, id. x 30 cm) packed with 300 mg of activated charcoal. If a precipitate was present in the urine it was removed by centrifugation.
The charcoal was washed with 30 ml of distilled water, an amount sufficient to prevent any white precipitate with a 10 per cent silver nitrate solution.
Glacial acetic acid was added until 10 ml of eluted solution was collected in the first tube (1.2 X 12 cm). The solution was evaporated within five minutes.
Two millilitres of water was added to the residue and then mixed; thereafter the pH of the solution was adjusted to 9.0 with about 0.5 ml of a 3N K 2CO 3 solution.
The solution was extracted by shaking with 5 ml of 10 per cent isopropanol in chloroform (10 per cent - IC) for five minutes. After centrifuging, 4 ml of the solvent was transferred to the second tube (2.1 X 10 cm), which contained 1 ml of N H 2SO 4. The solution was stirred for 20 seconds and then centrifuged.
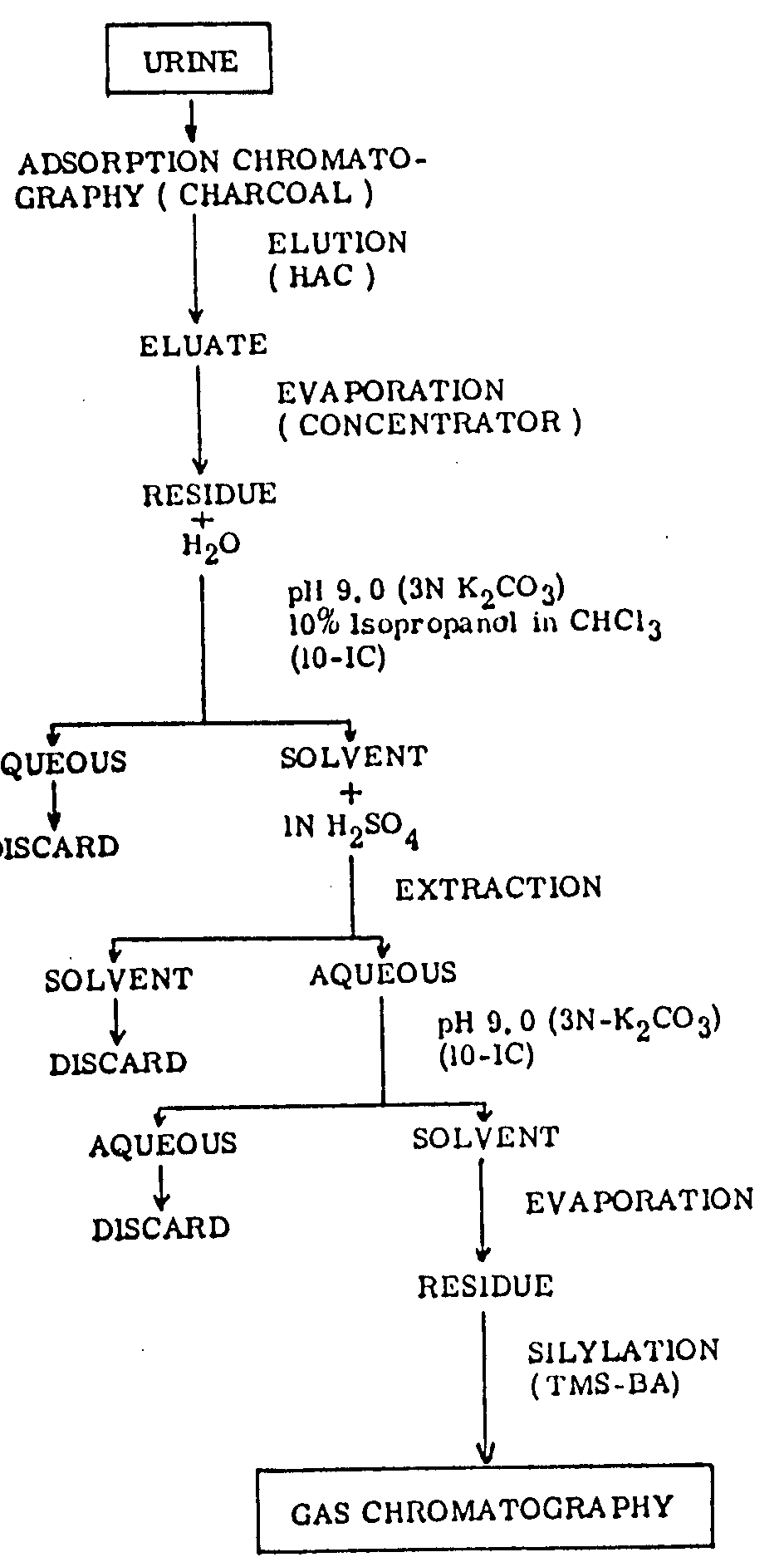
Nine-tenths of the acid layer was transferred to the third tube (2.1 X10 cm) which contained 200 mg of NaCl and the pH of the solution was adjusted again to 9.0 by adding 0.5 - 0.7 ml of a 3N K 2CO 3 solution.
Extraction was carried out by the same procedure as mentioned under 5 above, using 3 ml of 10 per cent - IC.
2.8 ml of the solvent was transferred to the fourth tube (2.1 X 10 cm) and evaporated. The residue was dissolved in 0.5 ml of methanol and pipetted into a GC-sample tube (0.7 X 5 cm).
After complete evaporation with the aid of nitrogen, the residue was dissolved in 50 µl of TMS-BA, placed on a hot plate at 60° for one minute.
The sample was left at room temperature for at least 20 minutes and then injected into the GC (figure I).
The residue from the extraction was dissolved in 50 µl of TMS-BA (25 per cent Bis-trimethylsilyl-acetamide in acetonitrile) by warming it in a hot water bath at 60 °C for a few minutes and then leaving it at room temperature for at least 20 minutes. A portion of this solution was thereafter used for GC analysis.
A Shimazu-5APF gas chromatograph equipped with a flame ionization detector was used. A very short glass column (6 mm od., 3 mm i.d. X 50 cm) was filled with solid Gas Chrom-Q, 80 to 100 mesh, coated with 3 per cent OV-17 (Applied Science Laboratories, Inc.). The temperatures at detection and injection port were 250° C and that of the column varied from between 90 to 220°, increasing at a rate of 6° C/min.
The applicability of the present method was tested by determining the excreted drugs in unchanged form, in rat urine. A drug was injected subcutaneously into four rats and their urine was collected during 24 hours after injection. The doses of the drugs used are shown in table 1.
Assay of each drug was done by comparison of the height of the recovery peak with that of each authentic standard.
|
Drugs |
Uncorrected |
Corrected |
|---|---|---|
|
MP
|
44.8 ± 0.50
|
68.8 ± 0.50
|
|
PE
|
71.2 ± 2.19
|
95.2 ± 2.19
|
|
MT
|
51.6 ± 1.36
|
75.3 ± 1.25
|
|
PZ
|
56.0 ± 2.67
|
79.9 ± 2.62
|
|
CC
|
56.4 ± 1.42
|
80.4 ± 1.46
|
|
CD
|
51.4 ± 3.05
|
75.4 ± 3.05
|
|
M
|
61.6 ± 1.51
|
85.6 ± 1.51
|
|
N
|
61.6 ± 3.73
|
85.6 ± 3.73
|
Values represent M ± S.E. from four determinations.
Fifty micrograms of each authentic standard was dissolved in 50 µl of TMS-BA; one microlitre of the solution was injected into the GC. The results are shown in figure II. Codeine was found to have the same retention time as that of morphine, but the peaks for the other drugs were clearly separated within 30 min. The peaks in figure III were obtained from urine to which 100 μg or each drug had previously been added. There is a clear correlation between these results and those shown in figure II. Hardly any barbiturates are extractable by this method but it is possible to detect them by modifying the procedure: barbiturates are extracted from the aqueous solution (pH 9.0) left after solvent extraction as described under 5 above, by adjusting the pH to 2.0.



The background for the chromatogram of human urine was examined with this procedure. Results are presented in figure IV. A GC sensitivity in the range of 64 x 10 2 allowed detection of drugs in amounts from 0.5 to 1.0 μg. For the drugs extracted, no disturbing peaks were found in the four samples; the second case which is shown to have a four times higher sensitivity than the other cases had a slightly higher background, about 10 per cent of the chart scale.
The efficiency of the method was tested by studying the percentage recovery of drugs added to urine; results are shown in table 1. Data were calculated by comparing the peak heights of extracted drugs with those of each standard. The percentage recovery was between 68.8 ± 0.50 and 95.2 ± 2.19 (corrected).
One does not imagine that all drugs will be detected at the same time in one urine sample. If one suspicious peak is found, isothermal conditioning procedures should be applied in order to shorten the retention time. The appropriate GC conditions for each drug and the limit of detection were tested and are given in table 2. Each drug could be detected in the range from 10-50 ng within 3.2 min. In such cases the range of sensitivity of the GC should be increased from 32 x 10 2to 4 x 10 2 just before the peak appears. The technique is described in detail by Yuji Maruyama and A. E. Takemori in "A new Gas Chromatographic Method for Estimation of Norepinephrine and Dopamine in Brain", Analytical Biochemistry 49: 240-247 (1972).
|
Temperature (T °C) |
||||
|---|---|---|---|---|
|
Drugs |
Inj. Det. |
Col. |
Rt. min. |
Det. Limit ng. |
|
MP
|
230 | 90 | 3.2 | 50 |
|
PE
|
180 | 150 | 2.8 | 20 |
|
MT
|
230 | 200 | 2.0 | 30 |
|
PZ
|
230 | 200 | 2.2 | 10 |
|
CC
|
260 | 205 | 2.0 | 20 |
|
CD
|
260 | 225 | 2.2 | 20 |
|
M
|
260 | 225 | 2.2 | 20 |
|
N
|
260 | 225 | 2.0 | 20 |
Sensitivity: 10 2 x 32 → 10 2 x 4.
The percentage recovery of each drug excreted in the urine in unchanged form was calculated and the results are shown in table 3. Data were simila1r to those reported by other investigators.
In conclusion, a simple procedure has been described for the extraction of several dependence-producing drugs from urine and a specific, sensitive and accurate gas chromatographic method has been established for the detection of these drugs. It has been demonstrated that 0.5 to 1.0 μg of an unknown compound could be satisfactorily detected at the primary screening by the temperature programming procedure and then the isothermal use of the assayed drug made it possible to detect the amount of 0.01 to 0.05 μg. Combination procedure of different conditions for GC was found to be practical for routine screening of an unknown compound.
|
Drugs |
Dose (mg/kg) s.c. |
Known recovery (%) |
Tested recovery (%) |
|---|---|---|---|
|
MP
|
10 | 5.5 | 7.3 |
|
PE
|
100 |
5-10
|
11.1 |
|
MT
|
20 | 10 | 14.5 |
|
PZ
|
40 |
some
|
1.5 |
|
CC
|
20 |
some
|
3.1 |
|
CD
|
30 | 2.8 | 1.0 |
|
M
|
20 |
5-10
|
21.7 |
|
N
|
30 |
5-10
|
11.7 |
D. J. Berry, J. Grove, B. Widdop and J. H. P. Willis, The detection of drugs of dependence in urine. Bull. Narc . XXII: 3, p. 31-37, 1970.
002S.J. Jule, M. L. Bastos, D. Jukofsky and E. Saffer. Routine identification of drugs of abuse in human urine. J. Chromatogr . 63, 289-301, 1971.
003J. M. Fujimoto and R. I. Wang: A method of identifying narcotic analgesics in human urine after therapeutic doses. Toxicol. appl. Pharmacol. 16 , 186-193, 1970.
004J. R. Broich, D. B. Hoffman, S. Andryauskas, L. Galante and U. Unberger: An improved method for rapid, large-scale thin-layer chromatographic urine screening for drugs of abuse. J. Chromatogr. 60 , 95-101, 1971.
005L. May and C. T. Kuo: Total analysis of an illicit or "street" narcotic sample by thin-layer chromatography. Bull. Narc . XXIV: 3, 35-36, 1972.
006N. H. Choulis. Separation and quantitation of mixtures of the most commonly abused drugs. J. pharm. Sci . 62:1, 112-114, 1973.
007S. H. Schnoll, R. D. Cohn and W. H. Vogel: A rapid thin layer chromatographic screening procedure for various abused psychotropic agents. J. psychedelic drugs, 5: 1, 75-78, 1972.
008L. T. Kenison, E. L. Loveridge, J. A. Gronlund and A. A. Elmowafi: Simplified multiple sample urinalysis in support of a methadone clinic. J. Chromatogr . (Amst.), 71: 1, 165-168, 1972.
009I.K. Ho, H. H. Loh and E. L. Way: Mini thin-layer chromatography in the detection of narcotics in urine from subjects on a methadone maintenance program. J. Chromatogr . (Amst.), 65: 3, 577-579, 1972.
010J.E. Wallace, J. D. Biggs, J. H. Merritt, H. E. Mailton and K. Blum: Sensitive thin layer chromatographic technique for determining morphine in urine. J. Chromatogr . (Amst.), 71: 1, 135-140, 1972.
011D.J. Beach and C. R. Angel: One system of mass drug screening. Scand. J. Clin. Lab. Invest ., 29 Suppl. 126: 15, 1972.
012Y. Maruyama and E. Hosoya: Studies on the fate of fentanyl. Keio J. Med ., 18, 59-70, 1969.
013Y. Maruyama. Studies on the distribution, the metabolism and the excretion of pethidine. I. Detection of pethidine and its metabolites in urine by thin-layer chromatography. Journal of the Keio Medical Society (in Japanese), 46, 55-59, 1968.
014E. Hosoya. Establishment of the method for the detection of small amounts of narcotics in human urine. Drug abuse, United States-Japan Cooperative Science Research Programme, 35-50, 1966-1968.
015Y. Maruyama, A. Sawa, A. E. Takemori and E. Hosoya. Gas chromatographic analysis of morphine and its glucuronide from urine. Fifth International Congress on Pharmacology, 23-28 July 1972, U.S.A.
016Y. Maruyama, Studies on the distribution, the metabolism and the excretion of pethidine. II. Quantitative determination of pethidine and its metabolites in urine by gas chromatography. J. Keio Med. Soc ., 46 , 55-59, 1968.

