INTRODUCTION
HISTORICAL
GENERAL PRINCIPLES
INSTRUMENTAL
PRESENTATION OF SPECTRAL DATA
SAMPLE HANDLING TECHNIQUES
INTERPRETATION OF SPECTRA
STUDIES ON ALKALOIDS
COLLECTIONS OF IR SPECTRA
APPRAISAL OF THE INFRARED METHOD
ADDITIONAL READING MATERIAL
Author: Charles E. Hubley , Leo Levi
Pages: 20 to 41
Creation Date: 1955/01/01
The Bulletin presents in this issue the continuation of the Canadian series of articles on the "Physical methods for the identification of Narcotics": Parts IVA and IVB, dealing with the infrared spectroscopic method. These articles describe the Solution and the Mull methods and the editors thought that it would be of interest to the readers to present in the same issue another method, evolved in 1953: the Pressed bromide method, so that a comparison could be made of the working of the three methods, as well as of the results obtained.
|
|
Page |
|
Introduction |
20 |
|
Historical |
20 |
|
General principles |
23 |
|
Origin of spectra |
23 |
|
Molecular vibrations |
23 |
|
Instrumental |
24 |
|
Energy sources |
24 |
|
Optical systems |
25 |
|
Radiation detectors |
25 |
|
Amplification of detector signals |
26 |
|
Measurement of I/I |
26 |
|
Presentation of spectral data |
26 |
|
Sample handling techniques |
26 |
|
Gases |
29 |
|
Liquids |
29 |
|
Solids |
29 |
|
Interpretation of spectra |
31 |
|
Basic considerations |
31 |
|
Qualitative analysis |
31 |
|
Quantitative analysis |
34 |
|
Studies on alkaloids |
34 |
|
Collections of IR spectra |
35 |
|
Appraisal of the infrared method |
35 |
|
Additional reading material |
35 |
|
References |
35 |
Infrared absorption spectroscopy as an analytical science has developed at such an accelerating pace during the past two decades that IR instruments are now very widely used in research, development and process control laboratories for qualitative and quantitative chemical analysis and for elucidation of molecular structure. It is the purpose of this paper to outline briefly some of the progress that has led to the present-day widespread use of infrared, to describe some of the more common methods of instrumentation and to indicate the scope and limitations of this very powerful analytical tool.
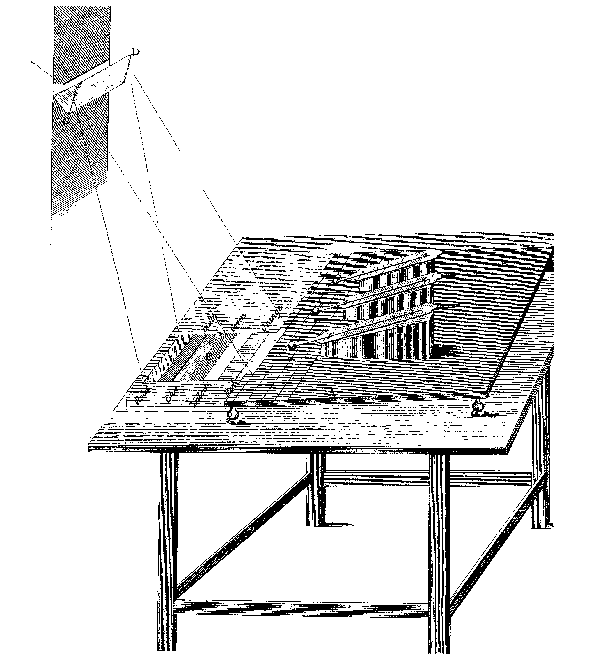
The roots of infrared spectroscopy extend to the beginning of the 19th century, when Sir William Herschel first demonstrated the existence of infrared radiation [1] 1. On placing the bulb of a sensitive thermometer in the various regions of the sun's spectrum dispersed by a glass prism, as illustrated in figure 1, he observed departures from the ambient temperature; a rise of 2o was noted in the violet, 3.5o in the green and 7o in the red. Since the brightest hue, green, was not that corresponding to the region of greatest temperature rise, Herschel sought the position of maximum energy and found it out beyond the red end of the spectrum.
There followed a few decades of controversy over whether the "infrared" energy was intrinsically the same as visible energy, or fundamentally different. Because the values obtained for reflection, refraction and transmission of energy by commonplace materials were not identical in the two regions, Sir William was erroneously led to believe that his was a new kind of radiation. However, the issue was later correctly resolved by his son, Sir J. F. W. Herschel, who proved to the satisfaction of the Royal Society [2] that, from the violet through to the infrared, the "new" energy was similar in nature to visible light. He showed that the sun's spectrum was continuous, except for the Fraunhofer regions (now known as regions of atmospheric CO 2 and H 2O absorption), and demonstrated this continuity by the differential evaporation of alcohol from a soot layer (see figure 2).
1. Numbers in parentheses refer to the list of references on pages 39, 40 and 41
An important milestone in the story of the alliance of IR spectroscopy and chemistry is the work of two of Her Majesty's Royal Engineers, Captain Abney and Lt.-Col. Festing ([3] ), reported in 1881. Operating in the very near infrared with glass optics and photographic plates they observed sufficient differences between the spectral lines produced by various alcohols, aldehydes, ethers, acids and esters to sense the possibility that future workers with improved apparatus might find a rich source of "clue(s) to the composition of a body". The vision of these early workers is best revealed by quoting their own words, "Weventure to think that the results we have obtained will prove that in their absorptions a still greater insight into the molecular constitutions of such bodies may be given. It seems to us that the spectra leave as definite characters to read as are to be found in hieroglyphics, and we venture to think that we have given a clue to enable them to be deciphered."

Infrared Red Violet
Basic contributions to the development of IR spectroscopy were also made by Young, Wallaston, Fraunhofer, Brewster, Talbot, Kirchoff, Fizeau, Fouchault, Seebeck, Becquerel, Nobili, Melloni, Svanberg, Langley, Rubens, Boys, Angstrom and many others. From their work came sensitive radiation detectors, sources, salt prisms and an improved understanding of the physics of electromagnetic radiation. The discoveries of some of these pioneers have been reviewed by Coblentz ([4] ).
In 1892 W. F. Julius ([5] ) first demonstrated that the presence of a methyl group in a molecule gave rise to characteristic IR absorption apparently independent of the constitution of the rest of the molecule. He made similar observations for amino, hydroxyl, aromatic and other functional groups, thus again drawing attention to the probable dependence of spectra on structure.
In 1905 W. W. Coblentz published his classic book Investigation of Infrared Spectra ([6] ). It contains the spectra of a large number of organic and inorganic compounds, including gases, liquids and solids, and relationships between spectra and structure are discussed in detail. Indeed, this book is still well worth study by the present-day spectroscopist, not only for its historical interest but also for its factual content. Though most of Coblentz's observations have since been re-examined and extended, virtually none has required contradiction.
During the next quarter century rapid strides were made along theoretical and experimental lines. By the mid-1930s it was realized that if the speed of recording IR absorption spectra could be substantially increased, industry would have a valuable tool for product control and research. With this goal in mind a few American companies added appropriate mechanical and electrical features to high-dispersion spectrometers and succeeded in developing automatic recording IR instruments for their own use.
During the war, when many critical production problems required rapid solution, instrument development was given federal support in several countries. As a result, there have emerged for general use in the postwar era many commercial types of recording IR spectrometers, all of which are capable of producing quickly spectra of high quality. Spectra which would have required eight hours of laborious point-by-point measurements by a skilled scientist in a carefully selected, dry, dark, vibrationless room thirty years ago can now be run off routinely in a few minutes by a laboratory technician using an instrument relatively insensitive to environmental conditions. Figure 3, showing a photograph of the spectrometer used by Coblentz ([6] ), illustrates a typical pioneer infrared laboratory. The temperamental nature of these early spectrometers is further exemplified by quoting from Abney's paper ([3] ), "A want of rigidity in our laboratory floor" caused spectral lines to be "irregularly-spaced and ill-defined", a condition which improved markedly when the apparatus was moved to " amore stable site".
For the information of those interested in delving more deeply into the fascinating history of infrared spectrometry, a bibliography of 2,700 references is contained in a 1944 publication by Barnes, Gore, Liddel and Williams ([7] ).

For the discussion that follows it will be useful to define a few common terms and symbols:
Transmittance: T = I/I o, where I o and I are the intensities of incident and transmitted radiant energy, respectively; i.e., the beam strength before and after sample absorption.
Micron(wavelength) : μ = 10 -6 metre = 10 -3 mm.
Wavenumber(frequency) : v = cm -1 = 1 / μ x 10 4, i.e., the number of waves per centimetre.
When infrared radiation of a given frequency encounters a molecule, energy is absorbed, provided the frequency of the incident radiation corresponds to a natural vibration frequency of the molecule. Therefore, when the values of I/I o for narrow bands of infrared radiation are plotted against spectral positions, in either frequency or wavelength units, a spectrum is obtained, which can often be interpreted in terms of structural characteristics of the irradiated sample.
To visualize the mechanism whereby the infrared radiation is absorbed one must consider that all the atoms of any molecule not at absolute zero are constantly oscillating about their equilibrium positions. The oscillation frequencies, which depend upon the masses of the atoms and groups involved and the bond forces between them, are of the same order of magnitude as the frequencies of near infrared radiation (5,000 to 500 cm -1).
Because every atom in a molecule has three directions of motion, any molecule of N atoms will have 3N degrees of freedom ([8] , [9] ). However, since there are three degrees of translational freedom, except for an adsorbed molecule, and three degrees of rotational freedom, except for a linear molecule, there are in general 3N-6 possible degrees of vibrational freedom. Whether or not all these degrees of freedom will appear as "fundamental modes" in an infrared absorption spectrum will depend on whether the vibrations cause periodic changes in dipole moment, i.e., cyclic separation of positive and negative charges. If no such changes in dipole moment occur there can be no interaction between the IR beam and the sample, and no absorption of energy. In such an instance the vibration is said to be "forbidden" in the infrared. As a simple example, the homonuclear diatomic gases, H 2, O 2, N 2, etc., may be chosen. Being linear these molecules lack one degree of rotational freedom and each has 3N-5=1 fundamental mode of vibration only. However, their symmetry prevents any periodic change in dipole moment, the absorption band corresponding to their normal vibration frequency is forbidden in the infrared, and hence these gases are transparent. For heteronuclear diatomic molecules, such as HCl, HF, etc., a strong absorption band corresponding to the one vibration frequency, no longer forbidden in the infrared, appears in the IR spectrum.
For polyatomic molecules the number of fundamentals increases almost linearly. Hence, one might expect that for a molecule with a dozen atoms the IR spectrum would be complex and that for a simple steroid, with say 60 atoms, the spectrum would be valueless, particularly since multiples, sums and differences of frequencies often appear in addition to fundamentals. However, for various reasons IR spectra of complex molecules may often be quite simple:
High symmetry forbidding many of the fundamentals.
Coincidence or near coincidence of fundamentals (degeneracy).
Weakness of fundamental bands.
Thus, the spectra of complex molecules, although still defying complete analysis, can be of great value for purposes of compound identification and illumination of structure.
More rigorous discussions of the origin of spectra are to be found elsewhere ([10,] [11] , [12] ).
Intramolecular vibrations may be considered as simple harmonic vibrations following Hooke's Law, so that the frequency of a diatomic group can be calculated from the formula:
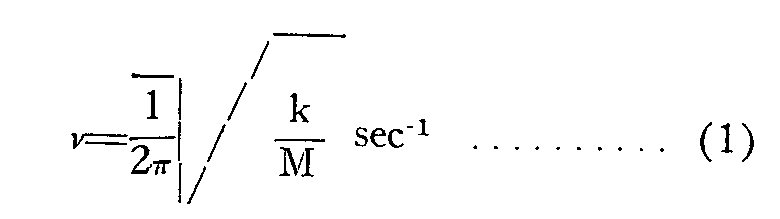
where v denotes the vibration frequency in cycles per second, k is the force constant and M the reduced mass of the atoms:
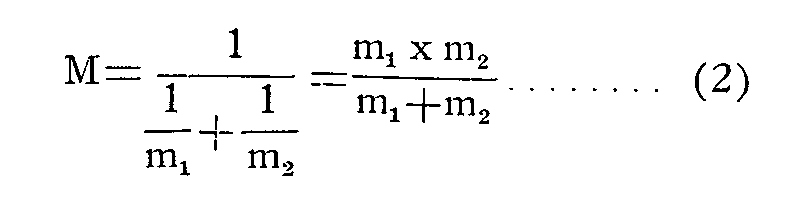
where m 1and m 2 are the masses of the two atoms, respectively.
It is useful to think of the force constant as the restoring force per unit extension or compression of the bond length, analogous to the ever-increasing "pull" or "push" one feels on moving one end of a spring from its equilibrium position.
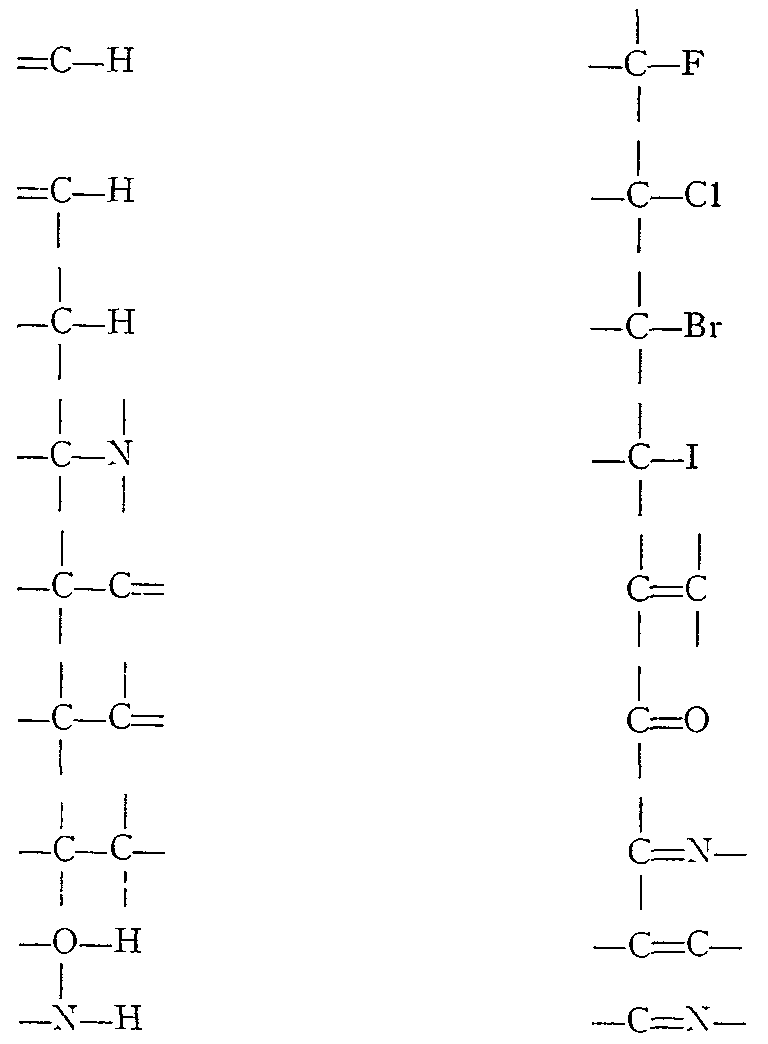
It will be seen from table I that for single bonds k usually lies between 4 and 6 x 10 5 dynes/cm., for double bonds, between 8 and 12 x 10 5 dynes/cm., and for triple bonds, between 12 and 18 x 10 5 dynes/cm.
Several general rules can be deduced from table I and the equations relating atomic masses, force constant and vibration frequency:
Increasing bond order, decreasing reduced mass or increasing bond strength moves the absorption band to a higher frequency (shorter wavelength) position.
The greater the electronegativity of atom X in a C-X bond, the higher the frequency at which C-X absorption will occur. This is illustrated in the decreasing value of the force constant in the series C-F, C C1, C-Br and C-I.
If the mass of one atom in a vibrating pair is much greater than that of the other atom, the reduced mass term in the frequency equation will be governed mainly by the mass of the lighter atom. Thus absorption bands for O-H, N-H, C-H, and S-H all appear in the same general region of the spectrum, namely between 2.7and 3.9 microns.
It is not possible to predict frequency values confidently from a priori considerations. Precise information regarding bond strengths is not always available nor can these be calculated because of unpredictable influences that neighbouring groups (extra- and intra-molecular) might have on bond characteristics. Therefore, it is generally more satisfactory to consult assignment tables and charts, such as those shown later in this paper (table III and figure 9), than to attempt calculating fundamental frequencies from inadequate data. However, when information on specific assignments is not available, a variation of equation 1 ([15] ) is sometimes useful for estimating where a specific band might be located:

In this equation K equals k X 10.5dynes/cm, and V is the reduced mass of the atom pair in dimensionless atomic weight units. As an illustration, the frequency of the carbonyl linkage in acetone may be calculated as follows:

This frequency lies within 10 cm -1of the observed value. To assist one in making computations of this type two empirical rules, by Badger ([16] ) and Gordy ([17] ), give mathematical approximations of force constants from other fundamental atomic data.
In addition to stretching vibrations involving periodic changes in bond lengths, molecules undergo other infrared active vibrations, involving bond angles. These are known as bending or deformation vibrations and are further classified as wagging, rocking or twisting to describe the direction of oscillation of bond angle relative to an axis or symmetry plane in the molecule ([14] ). It is the deformation frequencies which often are of greatest value in giving individuality to the spectra of isomers.
A spectrometer consists essentially of three parts - a source of energy, an optical system for directing and dispersing the energy and an energy detector. Each portion of the electromagnetic spectrum dictates a different choice of these parts but the infrared requirements are probably most exacting.
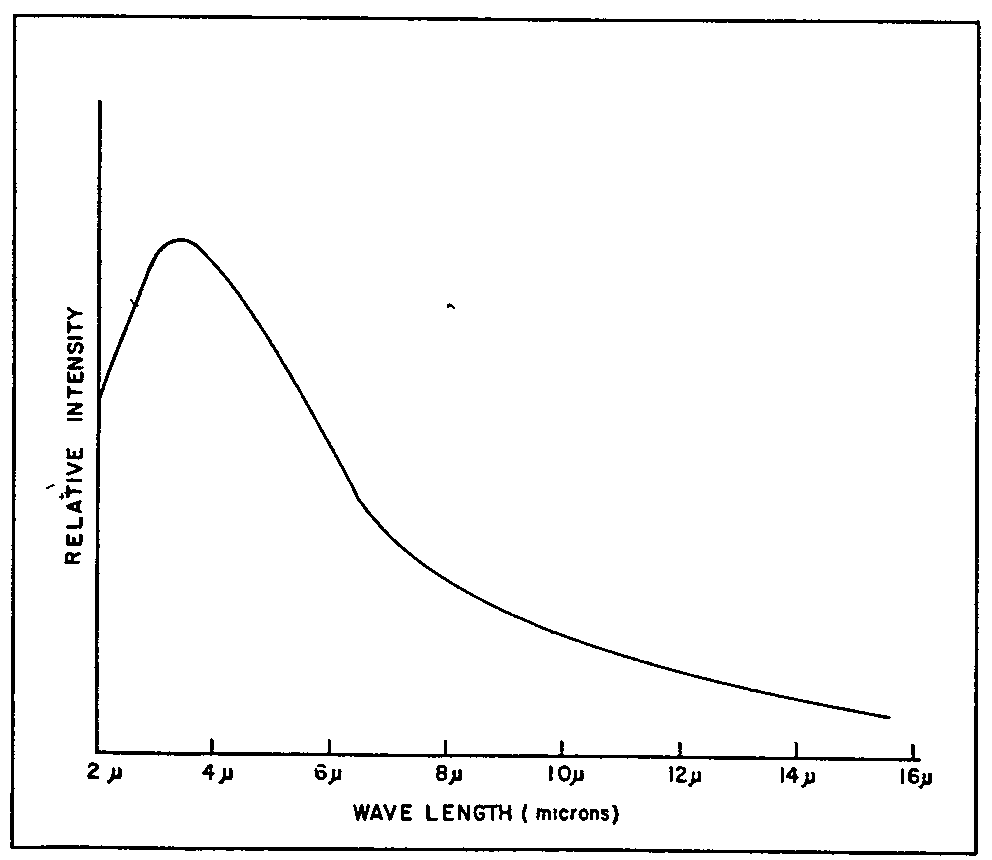
The frequencies of the vibrational infrared are lower than those of visible light yet higher by a factor of about 1,000 than the shortest radio waves. These infrared frequencies are generated by vibrating atoms, which process requires heat and materials able to withstand high temperatures. The most commonly used sources are the Nernst glower and the globar, although a tungsten filament lamp is sometimes employed in the very near (photoelectric) infrared.
The Nernst glower is a high emissivity source fabricated from a mixture of oxides, including those of zirconium, thorium and cerium. It is heated electrically and operated at temperatures above 1,500°C. because of the selective emission of the component metal oxides at lower temperatures in the very near infrared. On account of its large negative temperature coefficient of' resistance the Nernst glower must be heated to several hundred degrees before it becomes sufficiently conducting to attain incandescence at its normal operating-voltage.
The globar is an electrically heated rod of silicon carbide (carborundum). It needs no preheating. However, because of the high wattage at which it is normally operated, it requires water cooling.
Both sources give an essentially black-body type of emission curve but they differ somewhat in their relative energies at various wavelengths. In general, the globar gives more energy at longer wavelengths (relative to its short wavelength energy peak) than does the Nernst glower. Therefore, the choice between sources depends to some extent on the application.
A weakness common to both sources is the rapid decrease in output with increasing wavelength, as illustrated in figure 4. The very sensitive detectors required to pick up the faint longwave energy are unfortunately also activated by any stray energy from the intense shortwave region. The non-uniform source emission therefore often creates a very serious scattered light problem at the long wavelength end of the spectrum, a difficulty which has been substantially reduced in modern instruments, however, by optical or electrical filtering.
Because of the absorption of infrared energy by most matter the energy from the infrared source usually is directed through the spectrometer by front surfaced mirrors rather than conventional mirrors or lenses.
Dispersion can be accomplished by an echelette diffraction grating or a prism. Though the former is required in the very far infrared, and is used in the near infrared for extreme resolution, the latter has proved quite satisfactory for routine use. Moreover, the prism is simple to operate, gives reasonable dispersion, and has formerly been cheaper than a grating. With gratings now becoming reasonably priced, however, their popularity will undoubtedly increase ([18] ).
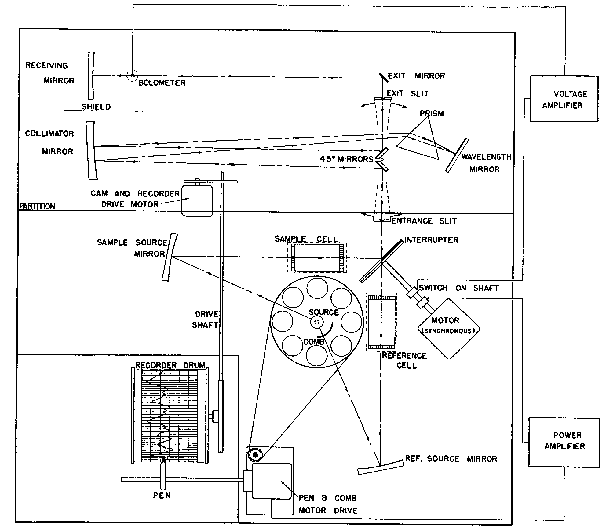
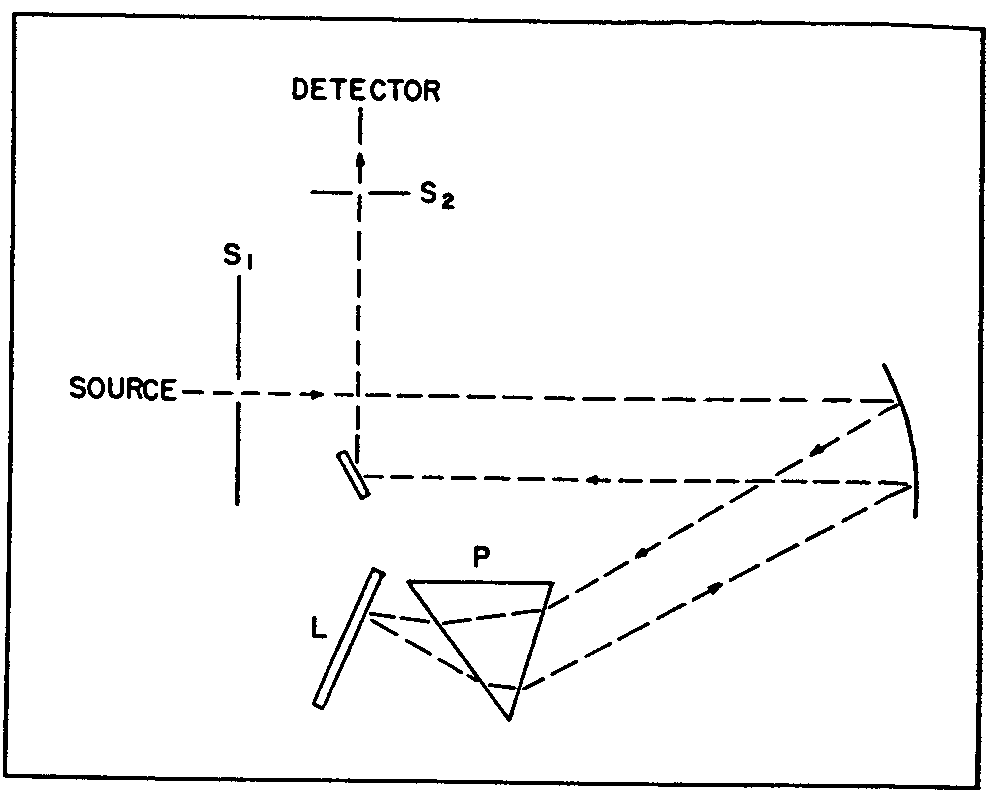
Most infrared prism spectrometers employ a monochromator in which the wavelength of the energy impinging on the radiation detector is determined by the angular position of a Littrow mirror. This type of monochromator is illustrated in figure 5, where S 1 is the entrance slit, P the prism, L the Littrow mirror and S 2 the exit slit. The energy reaching the entrance slit from the sample is polychromatic, while that emerging from the exit slit is monochromatic.
Although shortwave infrared radiation can be detected by means of the photoelectric effect (darkening of photographic paper or production of current in a photoelectric cell) it is the heating effect which is universally employed for this purpose at longer wavelengths. For detecting the low intensity radiation emerging from the IR monochromator, temperature sensitive devices are available which produce a usable electrical characteristic on being heated, e.g., bolometerswhich change in electrical resistance with temperature, and thermocoupleswhich generate an electric current. Either type will respond to temperature changes of as little as 10 -5 °C.
When acting as an infrared radiation detector, the bolometer forms one arm of a Wheatstone bridge. The unbalance of the bridge gives a voltage which is a function of the radiant energy falling on the bolometer and the "heating" current used to energize the bridge.
The bolometer element is generally a thin, narrow strip of metal. Its bulk is kept to a minimum since changes in its electrical resistance, i.e., its sensitivity, depend on temperature changes, which in turn will be maximal when the mass of the bolometer element is smallest. Similarly, if the bolometer is enclosed in an evacuated housing it will have enhanced sensitivity because heat will then be conducted from it more slowly.
The thermocouple consists merely of two bimetallic junctions, both of which are in the same environment except that only one is in the path of the IR beam. A temperature difference between the two junctions produces a voltage directly proportional to the intensity of the beam. As in bolometer design, the mass of the sensitive element of the thermocouple is kept small so that temperature changes and electrical effects are accentuated, and an evacuated jacket is used to increase sensitivity by reducing conductive heat loss.
Another type of heat detector, not as frequently used in commercial instruments as the above devices, is the Golay detector. It is a gas-filled cell, whose pressure is a direct function of the radiant energy falling on it. The pressure causes a thin, flexible cell wall to distort, and the extent of the distortion is then measured optically or electrically. Cells of this type are reputed to have a higher order of sensitivity than conventional bolometers and thermocouples ([19] ).
For transforming the very weak electrical signals from the radiation detector into electrical energy of recordable intensity high-gain, low-noise electronic amplifiers are now used. The amplified signals can operate a mechanical recorder giving a steady, reproducible record of the intensity of the radiation incident upon the detector. Because of the "zero" drift inherent in direct current amplifiers, a.c. amplifiers and chopped light beams (10-15 cycles per second) are quite generally used. A.C. amplifiers have the further advantage of narrow band pass tuning, which can eliminate unwanted signals such as those arising from electrical interference or mechanical vibration.
There are two basic methods by which the ratio of transmitted to incident energy can be measured.
The first is the "sample in-sample out" method whereby a spectrum taken with the sample in position is compared with that of the bank (i.e., no sample present). This is known as the single beam method and it requires that two spectra be run before the spectrum of the sample can be constructed. That construction of the final spectrum is a laborious operation can be readily appreciated when the complexity of the blank background spectrum of atmospheric CO 2 and H 2O is considered. Figure 6 shows this background and records the spectrum of a narcotic with and without such atmospheric background.
A more convenient method of obtaining the spectrum of a sample uses the "double beam" principle. Direct comparison of I and I° is made either by passing the energy from the source alternately through the sample and the blank to a single detector, or through the sample and blank simultaneously to independent detectors. In either case, appropriate electrical circuitry accomplishes the desired end, a direct record of I/I?. A typical instrument operating on the double beam in time (rather than in space) principle is illustrated in figure 7. Energy from the globar source is beamed in two directions, through the sample cell and reference cell, respectively. The two beams meet at the revolving semicircular mirrored sector, or interrupter, which alternately passes energy from each beam into the monochromator entrance slit. Radiation passing the entrance slit is then dispersed by means of the prism. The wavelength value of the beam passing through the exit slit, and hence reaching the bolometer, is determined by the angular position of the wavelength (Littrow) mirror. Because the interrupter permits the two beams (sample and reference) to impinge alternately on the radiation detector, any dissimilarity of the beams causes cyclic temperature fluctuations in the bolometer element and an alternating current in the bolometer circuit. The phase of this current (the polarity at any instant relative to the position of the revolving mirror) depends on which of the beams is the stronger, as does the polarity of the voltage from the mechanical rectifier monnted on the interrupter shaft. This voltage is fed through a d c. power amplifier and drives the pen and comb motor in the direction that alters the energy of the reference beam to match that of the sample beam. The comb, or optical density wedge, which moves in and out of the reference beam to maintain equality of the two beams, is geared to a recorder pen. By means of a mechanical linkage between the wavelength mirror and the recorder drum, an inked record of sample transmission versus wavelength (or frequency) can thus be obtained on chart paper as the spectral range of the instrument is scanned.
The issue of whether to record IR spectral data in frequency or wavelength units has often been the subject of inter-laboratory controversy. The relative merits of the two systems have been discussed by Jones ([20] ). In general, the frequency system is considered more satisfactory for correlation and interpretation, since its use facilitates the recognition of combination and overtone bands and permits direct comparisons to be made between IR and Raman data. Moreover, wave-numbers are directly proportional to energy, and the frequency of a band corresponds numerically and physically to the vibration frequency of a bond.
There have been a great many published examples of both qualitative and quantitative infrared applications using a wide variety of experimental techniques. Since nearly all of these are accessible through the references cited here and through standard scientific literature keys such as Chemical Abstracts, it is not proposed to attempt a comprehensive survey of techniques. Rather, it is intended to outline some of the more basic principles and indicate a few observations not normally mentioned, but which the authors have found to be particularly significant.
The most elementary requirements are that cell windows be reasonably transparent over the spectral region under investigation and that they do not react physically or chemically with the sample. To meet these requirements the materials listed in table II are available. The commonest window is NaCl since it is transparent throughout the most informative infrared region, is relatively cheap, is stable in the presence of most non-aqueous solutions and can be repolished by any experienced infrared worker ([21] )).
FIGURE 6
SPECTRUM OF (A) ATMOSPHERE AND (B) A NARCOTIC AS CHARTED BY A PERKIN ELMER DOUBLE BEAM MODEL 21 RECORDING INFRARED SPECTROPHOTOMETER ADJUSTED TO SINGLE BEAM OPERATION. THE SPECTRUM OF THE SAME COMPOUND (1,3-DIMETHYL-4-PHENYL-4-PROPIONOXYPIPERIDINE) AS TRACED BY THE INSTRUMENT WHEN IN DOUBLE BEAM OPERATION IS SHOWN IN (C)
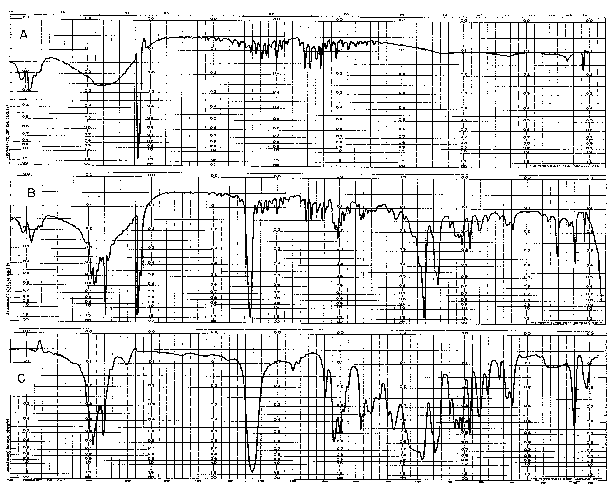
FIGURE 7
OPTICAL DIAGRAM OF TYPICAL DOUBLE BEAM SPECTROMETER
(Reproduced through the courtesy of Baird Associates, Inc., Cambridge, Mass.)
Table II
CHARACTERISTICS OF COMMON CELL WINDOW MATERIALS
|
Material |
Usable infrared wavelength range |
Remarks |
|---|---|---|
|
Quartz |
0.8 - 4µ |
non-hygroscopic |
|
CaF 9 |
0.8 - 10µ |
non-hygroscopic |
|
NaCl |
0.8 - 17µ |
hygroscopic |
|
KBr |
0.8 - 25µ |
hygroscopic |
|
0.8 - 25µ |
light sensitive |
In private correspondence with Dr. H. P. Schwarz, Chief, Division of Biochemistry, Philadelphia General Hospital, we have been told that the softness of AgCl windows causes them to scratch easily, so that even with very careful handling they change appreciably in transmittance from time to time. Dr. Schwarz also reports that a one-minute dip in dilute KCN solution, followed by a distilled water rinse, is a good conditioner (removes superficial scratches) for plates that have been used extensively.
bDr. H. J. R. Stevenson of the Department of Health, Education and Welfare, Public Health Service, Cincinnati, Ohio, in a private communication to the authors reports that badly scratched windows may be reconditioned by treating first with silicon carbide paper (240 to 400 grit), then with Buehler emery paper (0 to 000 grit), using kerosene instead of water as vehicle, and finally polishing on a disc covered with billiard cloth, employing a water suspension of grit 600 aluminium oxide during the operation.
Gas samples are particularly well-suited for infrared analysis. Vapour-tight cells are easily constructed of either glass or metal cylinders with appropriate windows cemented to the ends. For short path length cells (up to 10 cm.) the beam is routed directly through the cell containing gas at the pressure required to emphasize specific features of the spectrum. Gases usually display such a broad range of absorption intensities that it is common practice to run exploratory spectra at several different pressures. For weakly absorbing gas or gas from low vapour pressure liquids longer absorption paths are required. These are provided in multiple reflection cells where the path may be several metres even though the direct distance between entrance and exit windows is short enough for the cell to fit the confined sample space of a spectrometer. Several such cells, including some with variable path-length features, have been described elsewhere ([22] , [23] , [24] , [25] ).
Liquid samples are contained either in sandwich cells as contact films, or in cells of fixed or variable path length.
In making a contact film, a small amount of sample (one or two drops) is applied to an IR transparent window and spread into a thin film by placing another window on top. This sandwich technique is rapid and convenient for preparing liquid samples for IR examination. The films are of uncontrolled thickness, however, and are therefore used almost exclusively for qualitative work, the relatively uncommon exception being films of solutions containing internal indicators ([26] ) - - i.e., standard substances, present in known concentrations, and whose absorption band intensities can be related quantitatively to those of the compound being studied.
For most quantitative work cells of fixed path length are used. They consist of two windows separated by spacers of desired thickness, with one of the windows containing holes for admitting and removing sample. The authors have found liquid cells with path lengths as great as 15 mm. to be useful for some of their studies. Cells of 0.05-0.5 mm. path length are the most widely employed, however. They require a sample volume of about 1.0 cc. much of which is parasitic volume which does not intercept the energy beam. For most commercial instruments micro cells are also available which require only about 0.1 cc. of sample. Such cells are placed in the energy path at a position where the beam has a relatively small cross section, such as near the entrance slit (not applicable to optical null double beam instruments, which require that the sample be placed between the source and radiation chopper), near the source or in a position where the beam size has been reduced by a special lens system ([27] , [28] ). Within the past couple of years many suppliers of IR spectroscopic equipment have produced variable path length liquid cells, the sample depth in which can be altered at will by a micrometer adjustment. Such cells are particularly useful as compensating cells in double beam work since bands due to solvent absorption can be accurately cancelled from the recorded spectrum. Stuart ([29] ) describes a cell of this type embodying ruggedness and simplicity as basic design features. He also refers to variable thickness cells developed by other workers.
Solid samples often present the greatest challenge to the spectroscopist. The methods for handling them are numerous but all have their peculiar drawbacks, and it is often a delicate matter selecting a technique which will satisfactorily emphasize the spectral features required for the problem at hand.
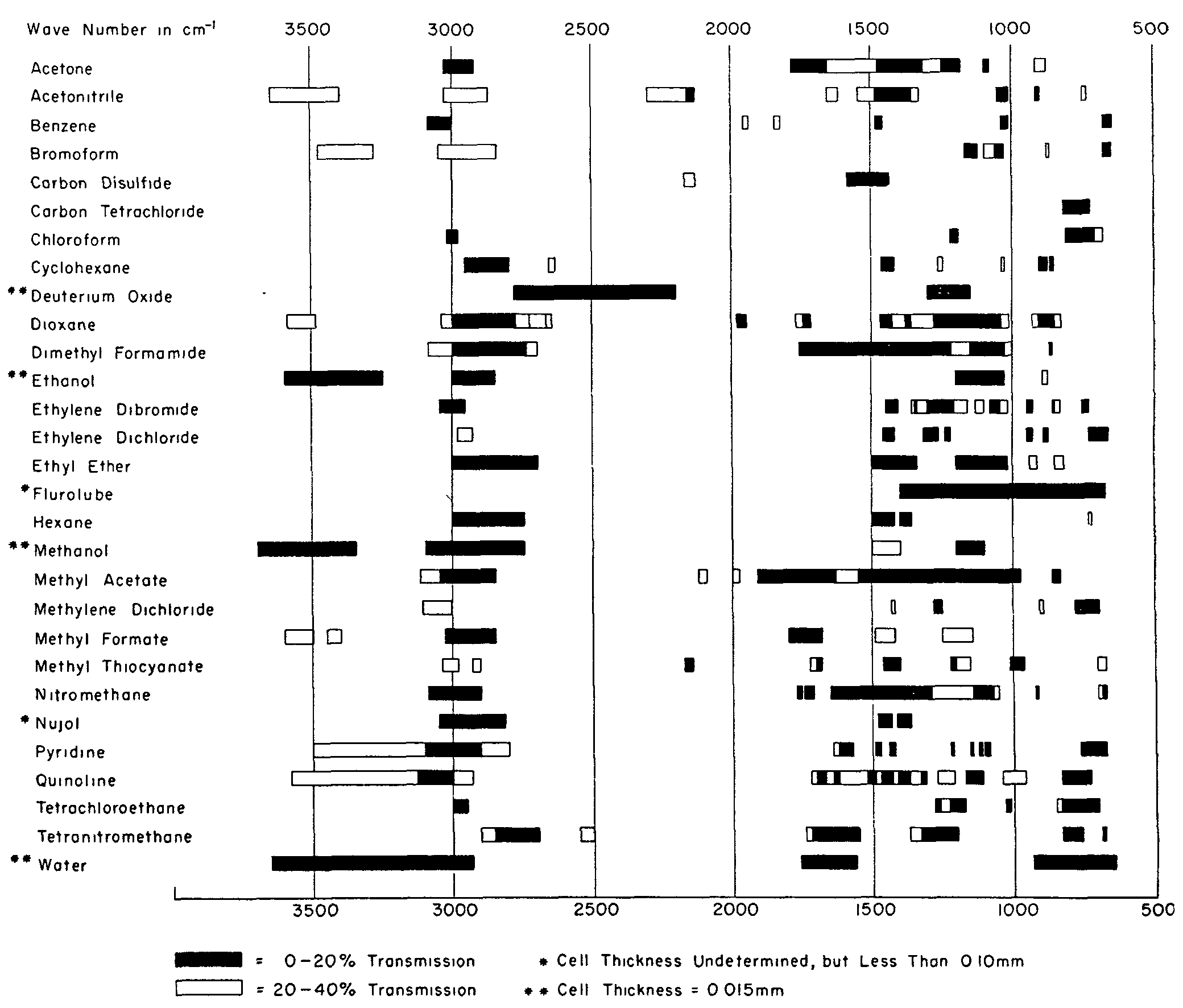
Solids may often be examined as solutions with quantitative precision. However, even with double beam instruments a false transmittance is recorded wherever the solvent absorbs strongly. Therefore, a complete and accurate spectrum can usually be obtained only by examining the solute in two or more solvents whose transparent regions are complementary. The problem of choosing suitable IR solvents has been formalized by others ([30] , [31] , [32] ) and, as a guide, transmission charts such as the one shown in figure 8 are available. All too often, however, the problem is not merely that of finding a desirable combination of solvents but, indeed, of finding a solvent at all.
For "insoluble" materials numerous ingenious methods have been devised. The two commonest will be discussed first and later some of the more specialized ones will be mentioned.
Mineral oil (e.g., Nujol) mulls have been the favourite solid sample preparation until recently. The sample and mulling agent are ground with mortar and pestle until a fine paste is obtained.2 The paste is then sandwiched between windows and examined. Because optimum sample thickness, concentration and particle size vary from sample to sample considerable skill is required to produce the best possible mull spectrum. A spectrum of this type is invalid wherever mulling agent absorption occurs (e.g., Nujol absorbs at the C-H wavelengths of 3.5, 6.9 and 7.3 µ). With other mulling agents, such as perfluoro nujol, perfluoro kerosene or halocarbons, the C-H absorption regions remain significant. However, since in both cases the sample thickness is unknown and not reproducible, the spectrum is only qualitative, unless an internal standard has been incorporated.
The method which is now largely replacing mulling is that of KBr pelleting ([34] , [35] ). It produces spectra which exhibit not only superior structural details but can also be evaluated quantitatively. Finely powdered KBr (other alkali halides can also be used) and samples are mixed in known proportions. The mixture is formed into a transparent disc or plate by means of a special dye which can be heated and evacuated ([36] ), both of which operations are sometimes required to promote transparency. The sample amount, required for quantitative interpretation, can be obtained either by weighing the disc or by gauging it with a micrometer.
2. The authors have found a stainless steel anvil and hammer powered by a hand vibratool ([33] ) to be a tremendous asset.
FIGURE 8
ABSORPTION CHARACTERISTICS OF IR SOLVENTS FOR CELLS OF 0.10MM THICKNESS
(Reproduced through the courtesy of R. C. Lord, Director, Spectroscopy Laboratory, Massachusetts Institute of Technology, Cambridge, Mass.)
Although the method has had a very popular reception by IR users, there are indications that it may not be applied indiscriminately to all systems. For example, the authors have found ([37] ) that barbiturate-pyridine-copper complexes showed less structure as KBr - than as mineral oil - spectra.
Since the introduction of the KBr method Sands and Turner ([38] ) have reported excellent quantitative results with solids incorporated into sheets of AgCl, mica and polyethylene, either by lamination or milling im- pregnation. Also, Dolinsky ([39] ) has described a method for obtaining quantitative spectra of finely pulverized solids dispersed in CS 2 or CCl 4 solution containing aluminium stearate.
Other techniques for handling solids include:
Mechanical film - a few materials, notably plastics and some minerals, can be run as thin films mounted on suitable holders.
Deposit on window - a deposit suitable for IR work may be produced by evaporating the solvent from a solution. It may be in the form of fine powder or a continuous film, depending on the nature of the material and the evaporation conditions ([40] ). It is desirable that the powder particles be of sub-micron size in order to prevent excessive scattering of incident energy within the spectrometer. A fine deposit can sometimes be attained by subliming or sputtering the sample onto a window, using an evacuated bell jar and an electrically heated sample boat ([41] , [42] , [43] , [44] ). This technique has been applied by Gettler, Umberger and Goldbaum ([45] ) for separating the components of mixtures of drugs and, in conjunction with infrared, it should prove a powerful tool to forensic chemists.
Melted film - it is sometimes possible with stable, low-melting materials to form a liquid contact film by heating the sample between IR windows. On cooling, the film solidifies and its absorption spectrum can be determined.
Partial thermal decomposition - the liquid, gaseous or solid thermal decomposition products of a sample can be compared with those of a known material, a technique which has found valuable application in instances where the solids could not be handled by any of the more standard methods ([46] ).
Many helpful discussions on IR experimental techniques exist, the most useful of these being in references [21] , [47] ,[48] and [49] .
The cardinal value of IR absorption spectroscopy lies in the uniqueness of the spectrum of any particular compound. It is this feature which determines the usefulness of the method for both qualitative and quantitative analysis. However, spectra are not of themselves articulate and therefore require interpretation. Moreover, spectra in the infrared are so strongly influenced by variations in instrument operating conditions, sample handling procedures, cell characteristics etc., that their interpretation is frequently complicated by these factors.
To aid in diagnosis of structure from spectra a number of useful group and bond assignment charts and tables have been prepared ([13] , [14] , [50] , [51] ). Table III and figure 9 are reproduced from two of these references ([13] , [50] ). The value of such guides cannot be overstressed, although it is well known by workers in the infrared field that the assignments are compromise values only, and hence should be applied with caution in analysing the spectrum of unknown material. It often happens that even for identification work a series of spectra of close chemical relatives of the unknown is required for comparison, while for quantitative work there is the additional requirement that all reference spectra be obtained on the same spectrometer as the unknown, and under identical operating conditions.
Successful qualitative interpretation of spectra requires a knowledge of the influences of particular structural features, sample environment, etc. Probably the most comprehensive source of information on interpretation of infrared spectra for use at the practical level is a very recent book by Bellamy ([52] ). It contains some 300 pages on absorption correlations, interpretations and assignments for most classes of organic and inorganic compounds, and discusses in detail the manner in which the absorption frequencies and intensities of specific functional groups are affected by such factors as conjugation, hydrogen bonding, ring strain, electronic influences of neighbouring atoms, etc. Since the book is also meticulously and exhaustively documented it will undoubtedly be a key reference work for those involved with interpretation of IR spectra.
Although there are other valuable formal treatises on qualitative interpretation (e.g., [50] , [53] ), it is proposed to review briefly some of the elementary principles of this field and discuss a few broad general features of IR spectra. The many characteristic group frequencies reported in table III have been obtained experimentally by making band position assignments for large numbers of spectra. By fortunate usage of such assignments it is often possible not only to determine that specific bonds, such as O-H, C-H or C=O, are present but also to deduce additional information about the bonds. For example, the free O-H band (2.7 µ) will shift to longer wavelengths (up to 3.0 μ) and broaden as the H becomes bonded to another atom, and the position of the C=O absorption suggests whether the carbonyl bond belongs to an ester, free acid or carboxyl ion, etc. Because of their value as detectors of specific functional groups spectra are becoming increasingly popular for monitoring chemical reactions, when sacrifice of the small quantity of material required for an IR spectrum can give strong evidence of whether and to what extent a reaction has gone in a particular direction. And, of course, if the corresponding spectrum is available for reference the presence of the end product can be established with a high degree of probability.
There is much other useful information that can be obtained from IR spectra. For instance, cis-trans isomers can often be differentiated because the trans form is normally more symmetrical and therefore less active in the infrared, as evidenced by fewer or weaker bands. In the special case of cis-trans isomers in which the mid-point of the C=C bond is also a centre of symmetry, the C=C vibration will be infrared inactive in the trans form and no absorption band corresponding to it will appear in the spectrum. Enol and keto tautomers in equilibrium mixtures can often be readily distinguished because of their different functional groups. Optical isomers which are mirror images (enantiomorphs) will have identical spectra when examined as gases, liquids or solutions. However, because of polymorphism they may often exhibit spectral differences in their solid spectra, which, in turn, will be different from those of the corresponding racemoids ([54] ). Diastereoisomers will have different spectra, regardless of sample phase.
FIGURE 9
SPECTRA-STRUCTURE CORRELATION CHART
PROBABLE POSITIONS OF CHARACTERISTIC INFRARED ABSORPTION BANDS
(Reproduced trought the courtesy of N. B. Colthup, Stanford Research Labratrories, Amerian Cyanamid Co., and the editor of the Journal of the Optical Society of America)
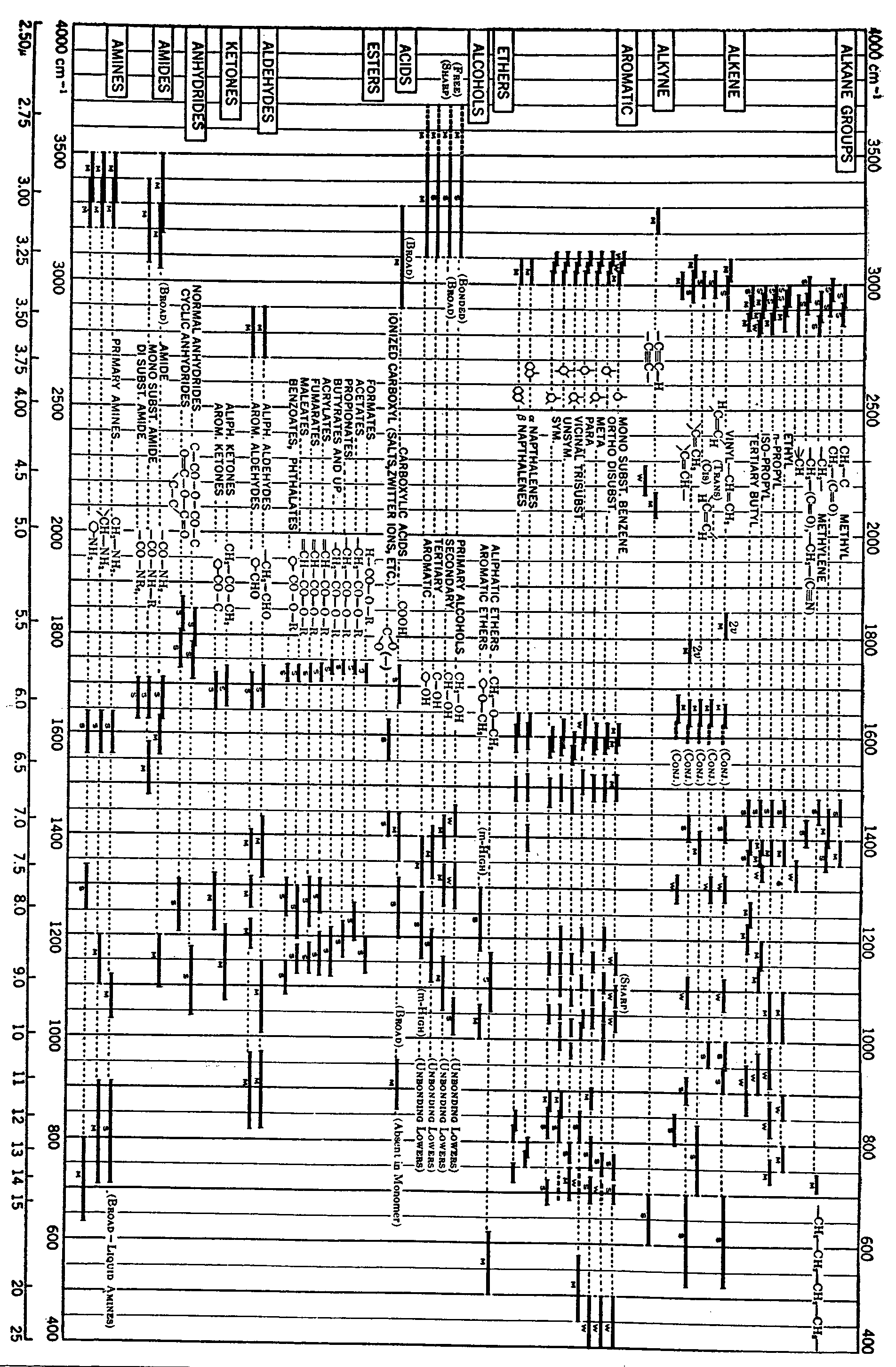
FIGURE 9
SPECTRA-STRUCTURE CORRELATION CHART
PROBABLE POSITIONS OF CHARACTERISTIC INFRARED ABSORPTION BANDS
(Reproduced trought the courtesy of N. B. Colthup, Stanford Research Labratrories, Amerian Cyanamid Co., and the editor of the Journal of the Optical Society of America)

Table III*
CHARACTERISTIC INFRARED ABSORPTION BANDS
|
R= alkyl group |
Ar = aryl group |
Ph = phenyl group |
~ =approximately |
|
(s)= strong |
(m)= medium |
(w) = weak |
(b) = broad |
|
Group |
Range (cm. -1) |
Range (μ) |
Reference | |
|---|---|---|---|---|
|
1. |
O-H |
|||
|
A. Stretching frequencies |
||||
|
-O-H (free) |
3730-3520 |
2.68-2.84 |
||
|
-O-H (associated) |
3520-3100 (b) |
2.84-3.22 (b) |
||
|
B. Deformation frequencies |
1080-1030 |
9.26-9.71 |
||
|
2. |
N-H |
|||
|
A. Stretching frequencies |
||||
|
- NH 2 (free) |
3550-3420 |
2.82-2.92 |
||
|
3450-3320 |
2.90-3.01 |
|||
|
N-H (associated) |
3500-3100 (b) |
2.86-3.23 (b) |
||
|
=N-H |
3400-3300 |
2.94-3.03 |
||
|
B. Deformation frequencies |
||||
|
-NH 2 |
1645-1550 |
6.08-6.45 |
||
|
-NH- |
1580-1510 |
6.33-6.62 |
||
|
3. |
C-H |
|||
|
A. Stretching frequencies |
||||
|
=C-H (acetylenic) |
3310-3200 |
3.02-3.12 |
[e] | |
|
=CH 2 (olefinic) |
3080 ±10 |
3.25 ±0.01 |
||
|
| |
||||
|
=C-H (olefinic) |
3020 ±10 |
3.31 ±0.01 |
||
|
Ar-H |
3090-3000 |
3.24-3.33 |
||
|
CH 3-C |
2960+-15 |
3.36-3.39 |
||
|
2870 ±5 |
3.48-3.49 |
|||
|
-CH 2- |
2926 ±5 |
3.41-3.43 |
||
|
2850 ±5 |
3.50-3.52 |
|||
|
| |
||||
|
-C-H |
2890 | 3.46 | ||
|
| |
||||
|
B. Deformation frequencies |
||||
|
-CH 2- |
1475-1425 |
6.78-7.02 |
||
|
-CH=CH 2 (see also C=C) ... |
1420-1395 |
7.04-7.17 |
||
|
CH 3-C1 |
1375 ±10 |
7.27 ±0.05 |
||
|
-CH 3 of isopropyl |
1380 (w), |
7.25 (w), |
||
|
1370 (w) |
7.30 (w) |
|||
|
-CH 3 of t-buty1 (see also C-C) |
1380 (w), |
7.25 (w), |
||
|
1370 (s) |
7.30 (s) |
|||
|
4. |
S-H |
~2580 (w) |
~3.88 (w) |
[g] |
|
Si-H |
~2240 |
~4.46 |
[h] | |
|
5. |
Deuterium |
|||
|
All D stretching frequencies (O-D, N-D, C-D, S-D, etc.) d 0.71 x corresponding H stretching frequencies (in cm. -1). |
||||
|
6. |
C=C |
2250-2150 |
4.44-4.65 |
|
|
H-C=CR |
2140-2100 |
4.67-4.76 |
[i] | |
|
R 1-C=C-R 2 |
2260-2190 |
4.42-4.57 |
[i] | |
|
7. |
C=C |
|||
|
A. Stretching frequencies |
||||
|
=C=(1,2-dienoid |
2200-1960 (s) |
4.55-5.10 (s) |
||
|
C=C (unconjugated) |
1650-1600 |
6.06-6.25 |
||
|
C=C (conjugated) |
1610-1580 |
6.21-6.33 |
||
|
B. Also |
||||
|
990 ±5, |
10.10 ±0.05 |
|||
|
910 ±5 |
11.00 ±0.06 |
|||
|
CH 2=CH-G (G = functional group other than ester, amide, or nitrile) |
990, 926 | 10.10, 10.80 | ||
|
CH 2=CH-E(E=ester group) |
990, 812 | 10.10, 12.32 | ||
|
CH 2=CH 1R 2 |
890 ±5 |
11.24 ±0.06 |
||
|
CH 2=C< (Terminal double bond on ring) |
875 | 11 43 | ||
|
R 1CH=CHR 2 (trans) |
1325-1275 (m) |
7.55-7.85 (m) |
||
|
980-965 (s) |
1020-10.35 (s) |
|||
|
R 1CH=CHR 2 (cis) |
1410-1350 (m) |
7.10-7.40 (m) |
||
|
715-685 |
14.0-14.6 |
|||
|
R 1CH=CR 2R 3 |
840-800 |
11.9-12.5 |
||
|
C. Phenyl ring |
1625-1575 |
6.15-6.35, |
||
|
1520-1480 |
6.58-6.75 |
|||
|
(1) Benzoyl (Ph-CO-) |
1600, 1584 | 6.25, 6.32 | ||
|
(2) Substituted phenyl |
||||
|
( a) Monosubstituted |
760-740 |
13.2-13.5 |
||
|
( b) ortho-Disubstituted |
750-740 |
13.3-13.65 |
||
|
( c) meta-Disubstituted |
790-770 |
12.7-13.0 |
||
|
( d) para-Disubstituted |
830-810 |
12.0-12.3 |
||
|
( e) 1,2,3-Trisubstituted |
770-760 |
13.0-13.2 |
||
|
( f) 1,2,4-Trisubstituted |
815-800 |
12.3-12.5 |
||
|
( g) 1,3,5-Trisubstituted |
835-825 |
12.0-12.1 |
||
|
D. Naphthalenes |
||||
|
α-Naphthalenes |
800-780 |
12.5-12.8 |
||
|
780-755 |
12.8-13.2 |
|||
|
β-Naphthalenes |
855-830 (m), |
11.7-12.0 (m), |
||
|
830-800 (s), |
12.0-12.5 (s), |
|||
|
760-720 |
13.2-13.9 |
|||
|
8. |
C-C |
|||
|
-(CH 2) - (x ≥4) (Singlet in liquid, doublet in solid paraffins. Actually due to CH 2 deformation) |
740-720 |
13.5-13.9 |
||
|
(CH 3) 2CH- |
1170 ±3 |
8.55 ±0.02 |
[o] | |
|
1145 ±5 |
8.73 ±0.04 |
|||
|
(CH 3.) 3C- |
1250 ±2 |
8.00 ±0.02 |
[o] | |
|
1208 ±6 |
8.28 ±0.04 |
|||
|
C-CH(CH 3)-CH(CH 3)-C |
1140-1110 |
877-9.01 |
||
|
9. |
C=N |
2400-2100 |
4.17-4.76 |
[p] |
|
R-C=N |
2260-2240 (s) |
4.43-4.47 (s) |
||
|
Ar-C=N, R-C=-N (conj.) |
2240-2215 (s) |
4.47-4.52 (s) |
||
|
-S-C=N |
~2160 |
~4.63 |
||
|
R-N=C (isocyanide) |
2200-2100 |
4.55-4.76 |
[p] | |
|
10. |
C=N |
|||
|
-N=C=S |
~2100 |
~4.76 |
||
|
-N=C=N- |
~2100 |
~4.76 |
||
|
-N=C< |
1660-1610 |
6.02-6.21 |
||
|
11. |
C-N |
|||
|
C-N-C(C saturated) |
1150-1100 |
8.70-9.09 |
||
|
C-N-C(C unsaturated or aromatic) |
1330-1250 |
7.52-8.00 |
||
|
N-CH 3 |
1370-1310 |
7.30-7.63 |
||
|
12. |
C=O |
|||
|
A. Stretching frequencies4 |
||||
|
Anhydrides-CO-O-CO- |
1860-1800 |
5.38-5.56, |
||
|
1800-1750 |
5.56-5.71 |
|||
|
Acid chlorides (unconjugated) |
1850-1780 |
5.41-5.62 |
[q] | |
|
Azlactones |
1820-1810 |
5.49-5.52 |
[q] | |
|
Chlorocarbonate R O-CO-Cl |
1800-1770 |
5.56-5.65 |
||
|
Lactones, ν |
1800-1760 |
5.56-5.68 |
||
|
Lactones, δ |
Normal ester position |
|||
|
Esters |
1760-1720 |
5.68-5.81 |
||
|
Unconjugated |
1755-1735 |
5.70-5.76 |
[q] | |
|
Conjugated |
~1720 |
~5.81 |
[q] | |
|
Also other bands as follows: |
||||
| (1) Formates |
1185-1160 |
8.44-8.62 |
[r] | |
| (2) Acetates |
1245, 665-635, 615-580 |
8.03, 15.0-15.7 16.3-17.2 |
[r] | |
| (3) Propionates |
1275, 1200-1190 (s), |
7.84, 8.33-8.40 (s) |
[r] | |
| 1080, 1020, 810 | 9.26, 9.80, 12.3 | |||
|
(4) n-Butyrates |
1255, 1190, 1100 | 7.97, 8.40, 9.09 |
[r] | |
| (5) Isobutyrates | 1260, 1200, | 7.94, 8.33, |
[r] | |
| 1160 | 8.62, | |||
| 1080 | 9.26 | |||
| (6) Isovalerates | 1195 | 8.37 | ||
| (7) Phthalates |
1285-1265, |
7.78-7.90, |
||
|
1130-1110 |
8.85-9.01, |
|||
|
1075-1065 |
9.30-9.39 |
|||
|
|
Aldehydes |
1730-1675 |
5.78-5.97 |
|
|
Ketones |
||||
| (1) Unconjugated |
1720-1705 |
5.81-5.86 |
[s] | |
|
(2) α, β-Conjugated (inc- luding aryl ketones) |
1700-1665 |
5.88-6.00 |
[s] | |
|
(3) α, β, α' β'-Conjugated (including diaryl ketones) |
1670-1650 |
5.99-6.06 |
[s] | |
|
(4) All methyl ketones but acetone (not stretching frequencies) |
1460, 1420, | 6.85, 7.04 |
[r] | |
| 1370, 1170, | 7.30, 8.55, | |||
| 595 | 16.8 | |||
|
Carboxylic acids, -COOH (see also O-H) |
1740-1650 |
575-6.06 |
||
|
B. Nitrogen-containing carbonyl compounds |
||||
|
Amino acid ions, NH 3+RCOOH |
1750-1700 |
5.71-5.88 |
||
|
β-Lactam (strained ring carbonyl) |
1825-1750 |
5.48-5.72 |
||
|
Ureido |
1720-1670 |
5.81-5.99 |
||
|
Amides |
||||
|
(1) R-CO-NH 2 (see also N-H) |
1690-1650 |
5.92-6.06 |
[w] | |
|
1630-1620 |
6.13-6.17 |
|||
|
(2) R-CO-NHR (see also N-H) |
1680-1640, |
5.95-6.10, |
[w] | |
|
1570-1530 |
6.37-6.54 |
|||
|
(3) R-CO-NR 3 |
1650 | 6.06 |
[w] | |
|
C. Miscellaneous carbonyl-containing groups |
C=O stretching frequency plus the following |
|||
|
Benzoyl, Ph-CO- |
1600, 1584 | 6,25, 6.31 |
[u] | |
|
Acetate, CH 3-CO-O |
665-635 |
15.0-15.7 |
||
|
615-580 |
16.3-17.2 |
|||
|
Acetyl, CH 3-CO- |
615-580 |
16.3-17.2 |
||
|
13. |
C-O salts |
|||
|
Carboxylate ion, -COO - |
1630-1550, |
6.13-6.45, |
[q] | |
|
1465-1400 |
6.82-7.14 |
|||
|
CO 3= |
1470-1400 |
6.80-7.14, |
||
|
880-810, |
11.4-12.3, |
|||
|
730-675 |
13.7-14.8 |
|||
|
HCO 3- |
1680-1610, |
5.95-6.21, |
||
|
1465-1400 |
6.82-7.14 |
|||
|
1025-970, |
9.75-10.3, |
|||
|
880-810, |
11.4-12.3, |
|||
|
730-675 |
13.7-14.8 |
|||
|
14. |
C-O |
|||
|
-O-CH 3 |
1340-1280 |
7.46-781 |
||
|
Ethers, unsaturated =C-O-C- (including aryl eters) |
1260-1200 |
7.94-8.33 |
||
|
Ethers, saturated |
1150-1070 |
8.70-9.35 |
||
|
580-540 |
17.2-18.5 |
|||
|
Triglycerides |
~1240 (m) |
~8.06 (m), |
||
|
1170 (s) |
8.55 (s), |
|||
|
1110 (w) |
9.01 (w) |
|||
|
15. |
C-F |
|||
|
-CF 2-and -CF 3 |
1350-1200, |
7.41-8.33, |
||
|
1200-1080 |
8.33-9.26 |
|||
|
=C-F |
1230-1100 |
8.13-9.09 |
||
|
-C-F |
1120-1010 |
8.93-9.90 |
||
|
16. |
C-Cl |
730-630 |
13.7-15.9 |
|
|
C-Cl(overtone?) |
1510-1480 (w) |
6.62-6.76 (w) |
||
|
-CCl 2 - |
845-795,~620 |
11.8-12.6, |
||
|
~16.1 |
||||
|
17. |
C-Br |
525-475 |
19.0-21.1 |
|
|
18. |
Silicon-containing groups |
|||
|
Si-H |
~2240 |
~4.46 |
[h] | |
|
-Si-CH 3 |
1410, 1260 (s) |
7.09, 7.94 (s) |
||
|
-Si-C 2H 5 |
1460, 1410, | 6.85, 7.09 |
[y] | |
| 1375, 1240, | 7.27, 8.06, | |||
| 1010, 960 | 9.90, 10.4 | |||
|
-Si-Ph |
1590, 1490, | 6.29, 6.71, |
[y] | |
| 1430, 1190, | 6.99, 8.40, | |||
| 1120, 1030, | 8.93, 9.71, | |||
| 995 | 10.05 | |||
|
-O-Si- |
1100-1000 |
9.01-10.0 |
[z] | |
|
-Si (CH 3) 3 |
840, 755 | 11.9, 13.2 |
[z] | |
|
-Ci (CH 3) 2 - |
800 (s), 700 |
12.5 (s), |
[z] | |
| 14.3 | ||||
|
19. |
Sulfur-containing groups |
|||
|
S-H |
~2580 (w), |
~3.89 (w), |
[g] | |
|
1000-900 |
10.0-11.1 |
|||
|
>C=S |
1550-1410 |
6.45-7.09 |
||
|
-C-S |
700-600 |
14.3-16.7 |
[g] | |
|
-S-S- |
~500 (?) |
~20.0 (?) |
[g] | |
|
Sulfoxide -S→O |
1060-1030 (s) |
9.43-9.71 (s) |
[bb] | |
|
Sulfones - SO 2 - |
1350-1300, |
7.41-7.69 |
||
|
1160-1120 |
8.62-8.93 |
|||
|
Sulfonates R-SO 2-OR |
1200-1150 |
8.33-8.70 |
[aa] | |
|
SO 4= |
1530-1450 |
6.54-6.90 |
||
|
S=P |
610-570 |
16.4-17.5 |
||
| 20 |
Miscellaneous groups |
|||
|
-NO 2(nitro) |
2500-2400, |
4.00-4.17 |
||
|
1590-1540, |
6.29-6.49, |
|||
|
1380-1320 |
7.25-7.58 |
|||
|
-N-O- |
~1000 |
~10.0 |
||
|
NO 3-(nitrate) |
1420 1370, |
7.04-7.30, |
||
|
845-815, |
11.8-12.3, |
|||
|
740-715 |
13.5-14.0 |
|||
|
P=O |
1380-1320 |
7.25-7.58 |
||
|
Phosphites |
870 | 11.5 | ||
|
Phosphonates |
940 | 10.6 | ||
|
21. |
Compounds of interest Nujol |
2918, 2861, | 3.427, 3.495, | |
| 1458, 1378, | 6.589, 7.257, | |||
|
720 (w) |
13.89 (w) |
|||
|
Liquid water (very thin) |
~3430, |
~2.92 |
||
|
1650-1600 |
6.06-6.25 |
|||
|
Atmospheric water vapour |
~1944-1320 |
~5.14-7.58 |
||
|
(much fine structure) |
||||
|
Atmospheric CO 2 |
2367. 2336, |
4.225, 4.281, | ||
|
721 (w), |
13.87 (w), |
|||
|
667 (s) |
14.99 (s) |
Reprinted with kind permission of the author and publishers of F. A. Miller's, "Application of Infrared and Ultraviolet Spectra to Organic Chemistry", Gilman's Organic Chemistry, Vol. III, 1953, John Wiley and Sons, Inc.
1May shift if CH 3 is attached to an atom other than C.
2Aryl group sometimes shifts 910 band.
3Sometimes overtone at 1830-1805 cm.-1,5.46-5.54 μ
4These are lowered by conjugation and by hydrogen bonding, and are raised by ringstrain. See discussion.
GENERAL REFERENCES FOR TABLE III
aBarnes, Gore, Stafford, and Williams, Anal. Chem., 20, 402 (1948).
bThompson, J. Chem. Soc, 328 (1948).
cRandall, Fowler, Fuson, and Dangl, " Infrared Determination of Organic Structures", D. Van Nostrand Co., New York (1949), chapters 3 and 4.
dRasmussen, "Infrared Spectroscopy in Structure Determination and Its Application to Penicillin", in L. Zechmeister's Progress in the Chemistry of Organic Natural Products, Springer-Verlag, Vienna (1948), pp. 331-386.
SPECIFIC REFERENCES TO TABLE III
eFox and Martin (a) Proc. Roy. Soc. (London), A162, 419 (1937); (b) A167, 257 (1938); (c) A175, 208 (1940); (d) J. Chem. Soc., 318 (1939).
fRasmussen, J. Chem. Phys., 16, 712 (1948).
gTrotter and Thompson, J. Chem. Soc., 481 (1946).
hThompson, ibid., 289 (1947), especially p. 293.
iWotiz and Miller, J. Am Chem Soc., 71, 3441 (1949).
jRasmussen and Brattain, J. Chem Phys., 15, 120, 131 (1947); Rasmussen, Brattain, and Zucco, ibid, 15, 135 (1947).
kSheppard and Sutherland, Proc. Roy. Soc (London), A 196, 195 (1949)
lTheumann and Wall, Anal Chem, 21, 1161 (1949).
mWhiffen and Thompson, J. Chem. Soc., 268 (1945).
nThompson and Torkhington, Trans. Faraday Soc., 41, 246 (1945).
oSimpson and Sutherland, Proc. Roy. Soc. (London), A199, 169 (1949)
pSheppard and Sutherland, J Chem. Soc., 453 (1947).
qRasmussen and Brattain, J. Am. Chem. Soc, 71, 1073 (1949).
rThompson and Torkington, J Chem Soc, 640 (1945).
sCromwell, Miller, Johnson, Frank. and Wallace. J. Am. Chem. Soc., 71, 3337 (1949)
tJones. Dobriner, and co-workers, ibid, 70, 2024 (1948; 71, 241 (1949). Jones and Dobriner, Vitamins and Hormones, Academic Press, New York (1949), vol. 7, pp. 294-363.
uRasmussen, Tunnichff, and Brattain, J. Am. Chem. Soc., 71, 1068 (1949).
vHartwell, Richards and Thompson, J. Chem. Soc., 1436 (1948).
wRichards and Thompson, ibid., 1248 (1947).
xWright and Hunter, J. Am. Chem. Soc., 69, 803 (1947).
yYoung, Servais, Currie, and Hunter, ibid., 70, 3758 (1948).
zRichards and Thompson, J. Chem. Soc., 124 (1949).
aaSchreiber, Anal. Chem., 21, 1168 (1949).
bbBarnard, Fabian, and Koch, J. Chem. Soc., 2442 (1949).
Ramsay[55] has pointed out that in many instances (e.g., overlapping or coincident bands) a bond cannot be characterized by the position of the absorption maximum alone. He suggests that measurement of the intensity of the absorption might provide additional information about the nature of the absorbing groups, and discusses three integration and extrapolation methods by which their true molecular absorption can be estimated from the spectrum. The methods have been applied with success to carbonyl bands in the IR spectra of steroids [56] . In these studies the integrated absorption intensities have been found to give much more satisfactory structural correlations than maximal molecular extinction coefficients. For instance, carbonyl intensities were found to be additive when two or three carbonyl groups were present in the molecule. This absolute approach to absorption intensities, formerly ignored because of uncertain instrument errors, may well increase the usefulness of IR spectral data.
There are several cautionary details to bear in mind when evaluating IR spectral data qualitatively:
The absence of a band in an assigned region does not preclude the possibility of a particular bond being present since symmetry may prevent the bond from being infrared active, e.g., the absence of a C=C band in ethylene.
The presence of a band in an assigned region does not positively establish the presence of a particular bond or group. There are many reasons why a band could be present, e.g., overtone, combination band, a band shifted into the region by peculiar environmental effects, etc.
Although every compound has a unique spectrum, the differences between the spectra of some compounds (e.g., higher members of homologous series) may be so minute as to be beyond the discrimination of the instrument.
Solid spectra are influenced by crystal form and crystal orientation. Thus a compound which is polymorphic may have more than one spectrum. Also, details in the spectrum of a compound can vary with different orientations of the crystals. In both instances, however, unique spectra can be obtained by melting or dissolving the solids.
Solution and mull spectra must be regarded as suspect in all regions where strong vehicle absorption occurs.
The basis for quantitative infrared analysis is the Lambert-Beer Law, which gives the theoretical relationship between the amount of absorption and the amount of sample:
I/Io= 10-kcl or log 10 Io/I=kcl
where k is the extinction coefficient, c is the concentration of the absorber, l is the cell path length, and I/I o has the significance already assigned. Log 10 I o/I is known as the optical density, and for any particular sample cell and absorption band it is directly proportional to concentration.
Only for quantitative work of a fundamental nature must the cell length be accurately known. Otherwise, fixed path-length cells are used and the system is calibrated by plotting optical density against a series of concentration values. In dilute solutions it can be assumed that changes in solute concentration can be made without effecting any significant change in the number of solvent molecules in the beam. Therefore, in solutions containing only one solute, the concentration of an unknown can usually be measured with good accuracy provided a strong absorption band exists in a region where solvent transmittance is high. Quantitative analysis can also be applied to multicomponent systems, since the absorbance of a mixture of components at any given wavelength is equal to the sum of the individual absorbance values, and such analyses are relatively simple if there is good separation of the bands used to gauge the amount of each component. However, if significant overlapping of absorbance regions occurs, as is often the case, it becomes necessary to set up and solve as many simultaneous equations as there are components, and it is difficult in practice to attain the same order of accuracy as with simpler systems.
Frequently, there are apparent departures from Beer's Law. These may be due to one or more of several causes:
Interaction of solution components.
Intermolecular association. Departures from linearity from this cause are greatest at high concentrations of the associating component.
Too wide a slit width. Beer's Law is strictly applicable only to monochromatic energy, which does not obtain literally except at extremely narrow slits.
Scattered radiation from another part of the spectrum causing false energy levels to be observed.
In general, quantitative IR methods are used most advantageously with low (but not trace) concentrations. For example, a shift in the D 2O content of ordinary water from 0.016 to 0.019 per cent is quite detectable, using CaF 2liquid cells of about 0.3 mm. path length ([57] , [58] ).
Although there have been several papers ([59] , [60] , [61] , [62] , [63] , [64] ) dealing with applications of IR absorption spectroscopy to alkaloid studies it is not surprising, considering the complexity of the molecules concerned, that in most of these the spectral data have been used only for purposes of compound identification or detection of functional groups. In reference [61] , however, a systematic examination of 47 alkaloids in chloroform solution was reported and assignments were made for some of the principal functional groups, e.g., O-H, N-H and C=O.
Spectra of 85 narcotics and related alkaloids are contained in Part IVB of the present series of papers.
Mention has already been made of reference material in which large collections of spectra may be found ([7] , [14] , [32] , [52] , [53] ). Other collections, also of appreciable size, include the spectra of malonyl urea derivatives ([65] ), minerals and inorganic ions ([66] , [67] ), phosphorus compounds ([68] ), steroids ([69] ), polymers ([70] ), plasticizers ([71] ) and unsulfonated monoazo dyes ([72] ). This compilation of references is far from complete, but alludes to groups of spectra which the present authors have found to be of particular interest.
In addition to the references already cited, there now exist on this continent several more comprehensive collections of IR spectra which are available to the public. Among these are the following:
The American Petroleum Institute Research Project 44 collection, which now contains over 1,600 spectra, mainly of hydrocarbons.
National Bureau of Standards Catalog of IR spectra, a collection of spectra of various series of compounds, e.g., alcohols, fatty acids, esters, ethers, etc., printed on McBee Keysort punched cards, together with other physical data and literature references. The present authors have contributed 64 spectra of barbiturates to this collection.
Sadtler's "Catalog of Infrared Spectrograms" (S. P. Sadtler and Son, Philadelphia 3, Pa.), a private collection of some 3,000 spectra of miscellaneous compounds, available commercially on Keysort cards.
It should be noted that continuing efforts are being made to transfer literature-spectra references to IBM cards, in order to guide spectroscopists to the correct source of spectral data through references tuined up by automatic sorting machines. Several thousand of these cards have already been issued by ASTM.
It would be misleading to give the impression that IR spectroscopy can provide all the answers required by the analytical chemist or other users. Like any analytical technique it has regions in which it is supreme and regions in which it must be acknowledged a poor competitor.
In relation to other analytical tools IR and Raman spectroscopy may be considered strong allies of UV and visible spectroscopy. Although the experimental techniques for IR and Raman are quite different, and particular bands may sometimes appear in the Raman but not in the IR (and vice versa), both methods can be treated together for the purpose of this discussion since both types of spectra reveal similar information concerning molecular vibrations and rotations. Visible and ultraviolet may likewise be grouped together since they both give information about electronic transitions.
Rotation-vibration spectra almost invariably contain more bands than electronic spectra (indeed, many organic compounds are transparent to visible and ultraviolet radiation, but none to IR and Raman), and they are more sensitive to changes in molecular structure. Ultraviolet and visible spectroscopy do have certain advantages in technique, however. Thus, a wide range of solvents (polar and non-polar) can be used, there is no need to employ fragile hygroscopic cell windows, and the accuracy of quantitative work is generally greater because inherent energy characteristics of ultraviolet radiation permit the use of more sensitive detectors and narrower slits (more linear Lambert-Beer Law plots are obtainable over a greater range of absorbances). However, improved instrumentation, new cell materials and novel techniques are continually broadening the experimental domain in which infrared spectroscopy may be regarded as the most useful analytical tool.
Readers interested in following developments in the field of infrared spectroscopy may find excellent literature guides in the annual (now biennial) reviews published originally by Barnes and Gore ([73] ) and continued by Gore ([74] ), in Analytical Chemistry. Applied Spectroscopy also gives a comprehensive coverage of activities in the IR field.
aIn private correspondence with Dr. H. P. Schwarz, Chief, Division of Biochemistry, Philadelphia General Hospital, we have been told that the softness of AgCl windows causes them to scratch easily, so that even with very careful handling they change appreciably in transmittance from time to time. Dr. Schwarz also reports that a one-minute dip in dilute KCN solution, followed by a distilled water rinse, is a good conditioner (removes superficial scratches) for plates that have been used extensively.
bDr. H. J. R. Stevenson of the Department of Health, Education and Welfare, Public Health Service, Cincinnati, Ohio, in a private communication to the authors reports that badly scratched windows may be reconditioned by treating first with silicon carbide paper (240 to 400 grit), then with Buehler emery paper (0 to 000 grit), using kerosene instead of water as vehicle, and finally polishing on a disc covered with billiard cloth, employing a water suspension of grit 600 aluminium oxide during the operation.
aBarnes, Gore, Stafford, and Williams, Anal. Chem., 20, 402 (1948).
bThompson, J. Chem. Soc, 328 (1948).
cRandall, Fowler, Fuson, and Dangl, " Infrared Determination of Organic Structures", D. Van Nostrand Co., New York (1949), chapters 3 and 4.
dRasmussen, "Infrared Spectroscopy in Structure Determination and Its Application to Penicillin", in L. Zechmeister's Progress in the Chemistry of Organic Natural Products, Springer-Verlag, Vienna (1948), pp. 331-386.
eFox and Martin (a) Proc. Roy. Soc. (London), A162, 419 (1937); (b) A167, 257 (1938); (c) A175, 208 (1940); (d) J. Chem. Soc., 318 (1939).
fRasmussen, J. Chem. Phys., 16, 712 (1948).
gTrotter and Thompson, J. Chem. Soc., 481 (1946).
hThompson, ibid., 289 (1947), especially p. 293.
iWotiz and Miller, J. Am Chem Soc., 71, 3441 (1949).
jRasmussen and Brattain, J. Chem Phys., 15, 120, 131 (1947); Rasmussen, Brattain, and Zucco, ibid, 15, 135 (1947).
kSheppard and Sutherland, Proc. Roy. Soc (London), A 196, 195 (1949)
lTheumann and Wall, Anal Chem, 21, 1161 (1949).
mWhiffen and Thompson, J. Chem. Soc., 268 (1945).
nThompson and Torkhington, Trans. Faraday Soc., 41, 246 (1945).
oSimpson and Sutherland, Proc. Roy. Soc. (London), A199, 169 (1949)
pSheppard and Sutherland, J Chem. Soc., 453 (1947).
qRasmussen and Brattain, J. Am. Chem. Soc, 71, 1073 (1949).
rThompson and Torkington, J Chem Soc, 640 (1945).
sCromwell, Miller, Johnson, Frank. and Wallace. J. Am. Chem. Soc., 71, 3337 (1949)
tJones. Dobriner, and co-workers, ibid, 70, 2024 (1948; 71, 241 (1949). Jones and Dobriner, Vitamins and Hormones, Academic Press, New York (1949), vol. 7, pp. 294-363.
uRasmussen, Tunnichff, and Brattain, J. Am. Chem. Soc., 71, 1068 (1949).
vHartwell, Richards and Thompson, J. Chem. Soc., 1436 (1948).
wRichards and Thompson, ibid., 1248 (1947).
xWright and Hunter, J. Am. Chem. Soc., 69, 803 (1947).
yYoung, Servais, Currie, and Hunter, ibid., 70, 3758 (1948).
zRichards and Thompson, J. Chem. Soc., 124 (1949).
aaSchreiber, Anal. Chem., 21, 1168 (1949).
bbBarnard, Fabian, and Koch, J. Chem. Soc., 2442 (1949).
Herschel, W.: "Investigation of the Powers of the prismatic Colours to heat and illuminate Objects; with Remarks that prove the different Refrangibility of radiant Heat. To which is added an Inquiry into the Method of viewing the Sun advantageously with Telescopes of large Apertures and high magnifying Powers", Philosophical Transactions, 90 , 255-326 (1800).
002Herschel, J. F. W.: "On the Chemical Action of the Rays of the Solar Spectrum on Preparations of Silver and other Substances, both metallic and non-metallic, and on some Photographic Processes", Philosophical Transactions, 130 , 1-59 (1840).
003Abney, W. De W.; Festing, E. R.: "On the Influence of the Atomic Grouping in the Molecules of Organic Bodies on their Absorption in the Infra-Red Region of the Spectrum", Philosophical Transactions, 172 , 887 (1881).
004Coblentz, W. W.: "Early History of Infrared Spectroradiometry", Science Monthly, 68 , 102 (1949).
005Julius, W. F.: "Bolometric Research on Absorption Spectra'', Verhandl. Akad. Wetenschappen Amsterdam, 1, 1-49 (1892).
006Coblentz, W. W.: Investigations of Infrared Spectra , Carnegie Institute, Washington, Publication No. 35 (1905).
007Barnes, R. B.; Gore, R. C.; Liddel, U.; Williams. V. Z.: Infrared Spectroscopy, Industrial Applications and Bibliography, Reinhold, New York (1944).
008Dennison, D. M.: "The Infrared Spectra of Polyatomic Molecules; I", Rev. Modern Phys., 3 , 280-345 (1931).
009Dennison, D. M.: "The Infrared Spectra of Polyatomic Molecules; II", Ibid., 12, 175-214 (1940).
010Hertzberg, G.: Infrared and Raman Spectra of Polyatomic Molecules , Van Nostrand, New York (1945).
011Harrison, G. R.; Lord, R. C.; Loofbourow, J. R.: Practical Spectroscopy , Prentice-Hall, New York (1948).
012Nielsen, H. H.; Oetjen, R. A.: Infrared Spectroscopy from W. G. Berl's Physical Methods in Chemical Analysis -Volume 1, Academic Press, New York (1950).
013Colthup, N. B.: Spectra-Structure Correlations in the Infrared Region, J. Opt. Soc. Am., 40 , 397 (1950).
014Randall, H. M.; Fowler, R. G.; Fuson, N.; Dangl, J. R.: Infrared Determination of Organic Structures , Van Nostrand, New York (1950).
015Gore, R. C.: Infrared Spectra of Organic Thiophosphates, Discussions of the Faraday Society , No. 9 138 (1950).
016Badger, R. M.: A Relation between Internuclear Distances and the Force Constants of Diatomic Molecules, J. Chem. Phys ., 2, 128-131 (1934).
017Gordy, W.: A Relation between Characteristic Bond Constants and Electronegativities of the Bonded Atoms, Phys. Rev., 69 , 130-131 (1946).
018Cole, A. R. H.: Performance of Merton-N.P.L.. Gratings in a Commercial Infrared Spectrometer, J. Opt. Soc. Am., 44 , 741 (1954).
019Bell, E. E.; Buhl, R. F.; Nielsen, A. H.; Nielsen, H. H.: Comparative Studies of the Performance of Infrared Receivers, J. Opt. Soc. Am., 36 , 355 (1946).
020Jones, R. N.: The Plotting of Infrared Spectra, Applied Spectroscopy, 6 (1), 32 (1951).
021Lord, R. C.; McDonald, R. S.; Miller, F. A.; Notes on the Practice of Infrared Spectroscopy, J. Opt. Soc. Am, 42 , 150 (1952).
022White, J. U.: Long Optical Paths of Large Aperture, J. Opt. Soc. Am, 32 , 285 (1942).
023Kratz, H. R.; Mack, J E.: An Improved Method for Obtaining a Long Optical Path in Limited Space, Phys. Rev. 57 , 1059(a) (1940).
024Smith, H. D.; Marshall, J. K.: Method for Obtaining Long Optical Paths, J. Opt. Soc. Am., 30 , 338 (1940).
025Lord, R. C.; McDonald, R. S.; Miller, F. A.: op. cit. , p. 154.
026Barnes, R. B.; Gore, R. C.; Williams, E. F.; Lindsey, S. G.; Petersen, E. M.: Infrared Analysis of Crystalline Penicillins , Anal. Chem., 19 , 620-627 (1947).
027Blout, E. R.; Bird, G. R.: Infrared Microspectroscopy; II, J. Opt. Soc. Am., 41 , 547 (1951).
028Anderson, D. H.; Miller, O. E.: Silver Chloride Beam Condensing Lens System for Micro Infrared Measurements , ibid., 43 , 777 (1953).
029Stuart, A. V.: Simple Variable Thickness Cell for Liquids, J. Opt. Soc. Am., 43 , 212 (1953).
030Jones, R. N.; Lauzon, R.: The Selection of Solvents for Infrared Spectrometry, National Research Council Bulletin No. 3, Ottawa, Canada (1953).
031Torkington, P.; Thompson, H. W.: Solvents for Use in the Infrared, Trans. Faraday Soc., 41 , 184 (1945).
032Pristera, F.: Solvents and Techniques in Infrared Spectroscopy, Applied Spectroscopy, 6 (3), 29 (1952).
033Ard, J. S.: Grinders for Mulling Infrared Microsamples, Anal. Chem., 25 , 1780 (1953).
034Schiedt, U.; Reinwein, H.: Zur Infrarot-Spektroskopie von Aminosäuren. Eine neue Präparationstechnik zur Infrarot-Spektroskopie von Aminosäuren und anderen polaren Verbindungen, Zeitschrift für Naturforschung, 76 , 270 (1952).
035Stimson, M. M.; O'Donnell, M. J.: The Infrared and Ultraviolet Absorption Spectra of Cytosine and Isocytosine in the Solid State, J.A.C.S., 74 , 1805 (1952).
036Hausdorff, H.: An Evacuable Die for the Pressed Potassium Bromide Technique, Applied Spectroscopy, 8 (3), 131 (1954).
037Levi, L.; Hubley, C. E.: Unpublished data.
038Sands, J. D.; Turner, G. S.: New Development in Solid Phase Infrared Spectroscopy, Anal. Chem., 24 , 791 (1952).
039Dolinsky, M. J.: Technique for Infrared Analysis of Solids Insoluble in Non-polar Solvents, J. Assn. Off. Agr. Chem., 34 , 749-763 (1951).
040Hacskaylo, M.: Preparation of Compounds for Infrared Spectrometry, Anal. Chem., 26 , 1410-12 (1954).
041Blout, E. R.; Fields, M.: The Infrared Spectra of Some Nucleic Acids, Nucleotides and Nucleosides, J. Biol. Chem., 178 , 335-343 (1949).
042Tyler, J. E.; Ehrhardt, S.: Infrared Spectra of Evaporated Films, Anal. Chem., 25 , 390 (1953).
043Wagner, E. L.; Hornig, D. F.: The Vibrational Spectra of Molecules and Complex Ions in Crystals; III. Ammonium Chloride and Deutero-Ammonium Chloride, J. Chem. Phys., 18 , 296-304 (1950).
044Wagner, E. L.; Hornig, D. F.: The Vibrational Spectra of Molecules and Complex Ions in Crystals; IV. Ammonium Bromide and Deutero-Ammonium Bromide, ibid., pp. 305 -312
045Gettler, A. O.; Umberger, Ch. J.; Goldbaum, L.: Fractional Sublimation on a Removable Transparent Film, Anal. Chem., 22 , 600-603 (1950).
046Harms, D. L.: Identification of Complex Organic Materials, Anal. Chem., 25 , 1140-55 (1953).
047Faraday Society Symposium on "The Application of Infrared Spectra to Chemical Problems, Trans. Faraday Soc., 41 , 171-297 (1945).
048Faraday Society Symposium on "Spectroscopy and Molecular Structure and Optical Methods of Investigating Cell Structure", Discussions of the Faraday Soc. , No. 9 (1950).
049Williams, V. Z.: Infrared Instrumentation and Techniques, Rev. Sci. Inst, 19 , 135-178 (1948).
050Miller, F. A.: Application of Infrared and Ultraviolet Spectra to Organic Chemistry, Gilman's Organic Chemistry , Vol. III, Wiley, New York (1953), pp. 143-150.
051Barnes, R. B.; Gore, R. C.; Stafford, R. W.; Williams, V Z.: Qualitative Organic Analysis and Infrared Spectrometry, Anal. Chem, 20 , 402 (1948).
052Bellamy, L. J. The Infrared Spectra of Complex Molecules , Methuen, London (1954).
053Rasmussen, R. S.: Infrared Spectroscopy in Structure Determination and its Application to Penicillin, Progress in the Chemistry of Organic Natural Products , Springer Verlag, Wien, (1948), p. 331.
054Eliel, E. L.; Kofron, J. T.: The Resolution of p-Ethyl-phenylmethylcarbinol. Infrared Spectra of Enantiomorphs and Racemates, J.A.C.S., 75 , 4585 (1953).
055Ramsay, D. A.: Intensities and Shapes of Infrared Absorption Bands of Substances in the Liquid Phase, J.A.C.S., 74 , 72 (1952).
056Jones, R. N.; Ramsay, D. A.; Keir, D. S.; Dobriner, K.: The Intensities of Carbonyl Bands in the Infrared Spectra of Steroids, ibid., p. 80.
057Patterson, W. A.: Private communication.
058Trenner, N. R.; Walker, R. W.: Infrared Determination of Deuterium in Liquid Water, Perkin Elmer Instrument News, 4 (1), 1 (1952).
059Adams, R.; Govindachari, T. R.: Senecio Alkaloids: α- and β- Longilobine from Senecio Longilobus, J.A.C.S., 71 , 1182 (1949).
060Gates, M.; Woodward, R. B.; Newhall, W. F.; Kungli, R.: The Synthesis of Ring Systems Related to Morphine; IV. N-Methylisomorphinane, ibid., 72 , 1141 (1950).
061Marion, L.; Ramsay, D. A.; Jones, R. N.: The Infrared Spectra of Alkaloids, ibid., 73 , 305 (1951).
062Bleat, G. B.; Harley, J. H.; Wiberley, S. E.: Use of Infrared Spectra in the Qualitative and Quantitative Determination of Alkaloids, J. Am. Pharm. Assn., Scientific Edition, 40(2), 107 (1951).
063Blout, E. R.; Fields, M.: Absorption Spectra; VIII. The Infrared Spectra of Purines and Pyrimidines, J.AC.S., 72 , 479 (1950).
064Eddy, C. R.; Eisner, A.: Infrared Spectra of Nicotine and some of its Derivatives, Anal. Chem., 26, 1432 (1954).
065Umberger, C. J.; Adams, G.: Identification of Malonyl Urea Derivatives-Infrared Absorption in Toxicological Analysis, Anal. Chem., 24 , 1309 (1952).
066Miller, F. A.; Wilkins, C. H.: Infrared Spectra and Characteristic Frequencies of Inorganic Ions, Anal. Chem., 24 , 1253-94 (1952).
067Hunt, J. M ; Wisherd, M. P.; Bonham, L. C.: Infrared Absorption Spectra of Minerals and other Inorganic Compounds, Anal Chem., 22 , 1478-1497 (1950).
068Daasch, L. W.; Smith, B. C.: Infrared Spectra of Phosphorus Compounds, Anal. Chem., 23 , 853-868 (1951).
069Dobriner, K.; Katzenellenbogen, E. R ; Jones, R. N.: Infrared Spectra of Steroids. An Atlas . Interscience Publishers, New York (1953).
070Hausdorff, H.: Analysis of Polymers by Infrared Spectroscopy , Paper presented at the Pittsburgh Conference on Analytical Chemistry and Applied Spectroscopy, March 7, 1951, and issued in Bulletin Form by the Perkin Elmer Corporation, Norwalk, Conn., U.S.A.
071Kendall, D. N.; Hampton, R. R.; Hausdorff, H.; Pristera, F: Catalog of Infrared Spectra of Plasticizers, Applied Spectroscopy , 7, 179-186 (1953).
072Dolinsky, M.; Jones, J. H.: The Infrared Spectra of Some Unsulfonated Monoazo Dyes, J. Assoc. Off. Agr. Chem. 37 (1), 197 (1954).
073Barnes, R. B.; Gore, R. C : Infrared Spectroscopy, Anal Chem., 21 , 7-12 (1949).
074Gore, R. C.: Infrared Spectroscopy, Anal. Chem., 22 , 750 (1950) ; 23, 7 (1951) ; 24, 8 (1952) ; 26, 11 (1954).