HISTORY
THE CHEMISTRY OF MORPHINE4
I. Known characteristics
II. Problems still requiring solution
CONCLUSION
Author: Paul B. WEILL,
Pages: 8 to 20
Creation Date: 1950/01/01

The knowledge of practices and remedies for conquering pain dates back to the earliest times. Although Babylon, Egypt and mythological Greece have left only rare and scanty evidence of their knowledge, Greece and Rome, on the other hand, have left us numerous detailed documents concerning analgesic preparations of acknowledged efficacy in those days.
These preparations, obtained mostly from plants, the most important of which were mandrake, henbane, the poppy and certain varieties of hemp, were completely described by Galen and Dioscorides, among others.
From the point of view of analgesic properties the opium poppy is certainly the plant with the widest and most justified reputation. Opium and its preparations, the knowledge of which was handed down through the Middle Ages by Arab medicine, have always been recognized as sovereign remedies against pain. Opium, as we know, is the dried juice obtained by making incisions in the green capsules of the opium poppy (Papaver somniferum), utilized varieties of which grow in the Balkans, Asia Minor, Persia, India, China, etc.
This raw product was used in medicine for centuries, in the form of tinctures, elixirs, compound powders, all of these being empiric forms whose activity differed according to their origin and concerning which neither the complex nature nor the fact that only one of the constituents was actually responsible for the activity was realized.
At the beginning of the nineteenth century, Sertürner[1] in Germany, Derosne[2] and Séguin in France thought out and accomplished the isolation of the active constituent of opium. This early work passed unnoticed and it was only the second publication of Sertürner, in 1817, in which for the first time this active principle was called "morphine", that aroused attention and gave rise to a large number of works.
This discovery was of considerable importance, because on the one hand it demonstrated the possibility of attributing to a clearly defined chemical product the activity of a complex drug and, on the other hand, a basic product had for the first time been isolated from a natural substance. The name "alkaloid" ("resembling an alkali") was adopted for this whole group of basic substances, large numbers of which were subsequently discovered.
In 1803, Derosne[1] had described as "salt of opium" a precipitate obtained by the action of potassium carbonate on an aqueous extract of opium; he had clearly observed its solubility in an acid medium and its precipitation by bases; but he had not emphasized either the novelty of the substance or its characteristics. This "salt of opium", according to Sertürner, was acid meconate of morphine or, according to Pelletier, narcotine, an alkaloid which was later isolated from opium but does not possess any narcotic activity, in spite of its name.
Another landmark was Séguin's communication to the Académie des Sciences, in 1804, announcing the isolation and characterization of morphine. As this work was not published until 1814, the discovery of the first of the alkaloids, morphine, is generally attributed to Sertürner.
Research, at first analytical, devoted itself to isolating the various constituents of opium, and establishing their formulae and constitution. This stage is not at an end, for the very structure of morphine still remains a matter of controversy. Assisted by the early progress made in the analytical field and by the tremendous impulse afforded by organic chemistry, synthetic work soon began. It was first directed towards the preparation of derivatives of the natural alkaloids of opium and later, indeed only recently, towards research into purely synthetic substances related to mor-phine by their analgesic properties and by a more or less remote similarity of composition.
All this work could neither have developed nor come to anything without the vigorous and fruitful efforts of pharmacologists and doctors. Important as it is, however, this dual pharmacological and clinical aspect of the question of morphine analgesics will here only be touched upon in passing, and solely in so far as it may help to clarify the question. The subject of the present article will be restricted to the chemistry of morphine, its derivatives and the synthetic analgesics.
Whatever their origin, the various types of opium seem to contain the same alkaloids, the only difference being in the proportions. Over twenty alkaloids have been isolated. Besides morphine they include some of its nearest related substances, such as codeine, thebaine, neopine; and others mainly belonging to a different chemical series, of which papavarine is the chief.
Only the first group of alkaloids chemically related to morphine will be considered here, since morphine cannot be studied without reference to the related alkaloids.
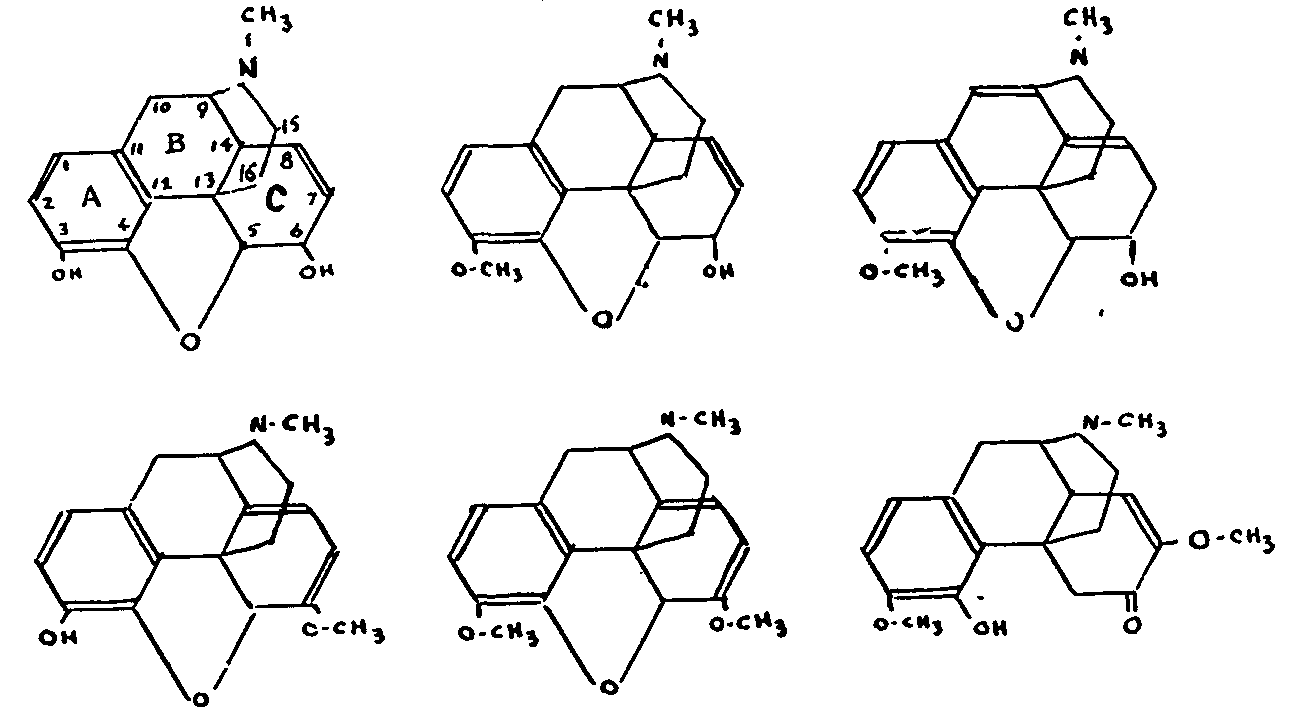
Before giving an account of the observations which have enabled us to build up our present knowledge of morphine, it seems necessary, for the sake of clarity, to start by giving the generally accepted formulae for the most important alkaloids in its group, which we shall have occasion to mention in the course of this article.
Of the six alkaloids represented below, only morphine and codeine are used in medicine. Neopine[5] is found in only very small proportions in opium. Oripavine[6] is present with its methyl ether, thebaine, in an Asiatic poppy, the Papaver orientale; its chemical composition was elucidated only in 1948. Thebaine, the most toxic of the opium alkaloids, is used as raw material for the preparation of derivatives such as eucodal and metopon. Although not found in opium, sinomenine is included here on account of its close chemical relationship to codeine. It was the subject of thorough research by Japanese writers, who described it as the optical antipode of a codeine derivative.
The observations or studies which brought to light the functional and structural characteristics on which the developed formulae of morphine and the related alkaloids are based are recalled below:
1. Elementary composition of the three main morphine alkaloids
Morphine: C 17H 19O 3N; codeine: C 18H 21O 3N; thebaine: C 19H 21O 3N
2. Presence in morphine of three oxygen atoms, one of which belongs to a phenolic hydroxyl (solubility in bases from which carbon dioxide re-precipitates it; change of morphine (phenolic) into codeine, or methylmorphine, by methylation; the second, to a secondary alcohol group (absence of the phenolic character of codeine; esterification; oxidation into a ketone, codeinone VII); and the third, chemically inert, to an epoxydic cycle (this point will be developed later).
Left to right:
MORPHINE
CODEINE
NEOPINE
Left to right:
ORIPAVINE
THEBAINE
SINOMENINE
3. Functional relations between morphine, codeine and thebaine
As we have seen, codeine is none other than methyl-morphine. Codeine itself is oxidized into codeinone, and the methyl ether of the enol form of this is the same as thebaine.
The latter alkaloid contains two methoxyl groups, as Zeisel's reaction shows, and two ethylenic bonds. The existence of these bonds was not easy to demonstrate: the catalytic hydrogenation of thebaine produces a complex mixture,[7] the main constituents of which are di-hydrothebaine VIII, and a ketonic base, dihydrothe-baine IX. It was not until 1927 that a compound identified as the dimethyl ether of morphine was finally isolated from the mixture of the products of hydrogenation.[8] To conclude this enumeration of the most striking chemical interrelations between morphine, codeine and thebaine, it should be recalled that, as Knorr demonstrated in 1909,[9] acids transform thebaine into codei-none VII.
Hence, after the demonstration of these close relationships between the three main morphine alkaloids, it was justifiable to apply to them as a whole the particular conclusions drawn from the structural study of one of them, a point which was very useful in elucidating their formulae.
4. Presence of a double ethylenic bond in morphine and codeine
This is easily demonstrated by hydrogenation of these alkaloids into their respective dihydro derivatives.[10]
5. Presence of a methylated tertiary-amine nitrogen atom
The basic properties of the morphine alkaloids are attributable to an amine function. In codeine, for instance, this is capable of fixing a single molecule of methyl iodide. The action of boiling potassium on this quaternary base produces a new tertiary base C 19H 25O 3N or methylmorphimethine. Repeating the same process (exhaustive methylation or Hofmann degradation) with the methylmorphimethine, it is possible to isolate, among other products of degradation, trimethylamine. The presence of this amine shows that one methyl group, and one only, is linked to the amine nitrogen of codeine: in the result, three methyls are fixed to the nitrogen in trimethylamine, whereas only two have been introduced during the two consecutive methylations effected.
6. The nitrogen atom of the morphine alkaloids forms part of a heterocycle
The degradation of codeine iodomethylate, described in paragraph 5, which is done without loss of nitrogen to form methylmorphimethine, already provides irrefutable proof of this. The study of the action of acetic anhydride on methylmorphimethine completes the demonstration, as the nitrogen atom is then detached in the form of dimethylethanolamine HO-CH 2-CH 2-N (CH 3) 2
As it has been shown that the molecule really separated was vinyldimethylamine, the nitrogen chain cannot be linked to the rest of the codeine molecule by oxygen (as this comes from the addition of water to vinylamine) but must be linked by the terminal carbon in its ethyl chain.[11]
7. Presence of a partially hydrogenated phenanthrene nucleus
The distillation of morphine on zinc powder had long indicated the presence of the phenanthrene nucleus[12] in the morphine molecule; but this too brutal reaction failed to show up the special characteristics of this phenanthrene. Phenanthrene oxygenated compounds[13] later identfiied by synthesis were obtained either by alcoholysis or acidolysis of morphine alkaloids, or by the Hofmann degradation of thebaine, methylmorphime-thine or its derivatives, fixing the positions of the sub-stituents in the morphine. Lastly, it should be noted that a partial saturation of the phenanthrene nucleus is necessitated by the hydrogen content of the morphine.
8. Positions 3 and 6 of the hydroxylic oxygen atoms and 4,5 of the epoxydic oxygen atom
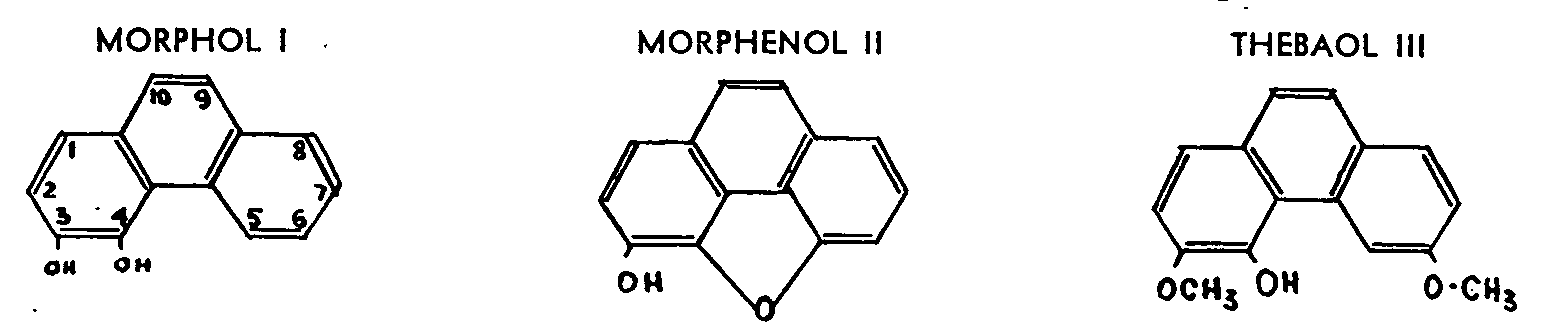
The action of acetic anhydride on the iodo-methylates of morphine or codeine, or directly on methylmorphi-methine, gives either morphol I[13] and morphenol II,[14] or their methylated derivatives, and thus makes it possible to fix the phenol group-free in the case of morphine and methylated in the case of codeine-in position 3.
The alcoholic hydroxyl is eliminated in the course of these degradations but survives in the case of thebaine, which gives thebaol III.[15] The fact that both thebaine and codeine can be transformed into the same ketone justifies the application to codeine of the conclusions drawn from the study of the degradation of thebaine, and hence the fixing of the alcoholic hydroxyls of codeine and morphine in position 6.
Moreover, since the action of acetic anhydride on codeinone makes it possible to isolate 3-methoxy-4:6 dihydroxyphenanthrene,[16] the respective positions of the phenol group and of the alcoholic hydroxyl must clearly be 3 and 6. The constitution of these phenanthrene derivatives (morphol,[17] morphenol, thebaol,[18] etc.) was explained by Pschorr, who pioneered in the field of phenanthrene synthesis and succeeded in synthesizing the substitute phenanthrenes which have made it possible to identify by direct comparison most of the products of degradation of the morphine alkaloids.
The acid dehydration of morphine and thebaine into apomorphine IV and morphothebaine V respectively fully confirms these results. Späth and Hromatka succeeded in synthesizing racemic apomorphine,[19] the benzoylated derivative of which is identical with the product of the benzoylation of levorotatory apomorphine.
9. Positions 7:8 of the double bond of morphine and codeine, position 6:7 and 8:14 of the two double bonds of thebaine
(a) Morphine and codeine
The action of chlorinating agents, such as PCl 3, PCl 5, or SOCl 2, on codeine gives α-chlorocodide, which may be transformed into β-chlorocodide or can be made by hydrolysis to yield a mixture of three codeine isomers. One of these isomers, isocodeine, is the result of a Walden inversion at C-6, as shown by its oxidation into codeinone VII and by the degradation of the latter into 3,4,6-trimethoxy-phenanthrene. The other two isomers, allo-codeine and allo-pseudocodeine, both oxidize into pseudocodeinone, the subsequent degradation of which gives 3,4,8-trimethoxy-phenanthrene VI.[20]
Hence it may be concluded that the four isomers form two pairs of stereo-isomers,-codeine and isocodeine, allo-codeine and allo-pseudocodeine-since neither of the pairs, on oxidation, gives more than one ketone, which is the same in each case.
The theory that the two pairs are linked by isomery goes back no further than 1923, when Gulland and Robinson,[21] and Wieland, independently advanced the hypothesis that an allyl migration took place, causing the alcoholic hydroxyl to move from C-6 to C-8, while the double bond shifted simultaneously from 7:8 to 6:7.
This migration falls into a well-known pattern-the same pattern as is used, in particular, to explain the geranoil-linalool isomery.
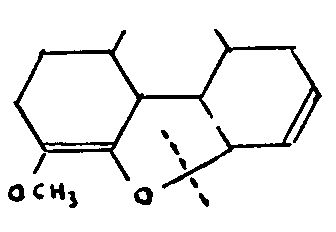
Still further confirmation is provided by the formation of phenol derivatives in the catalytic hydrogenation by hydrogenolysis of the allyl system of allo-codeinone and allo-pseudocodeinone, and the positions of the ethylenic bonds of morphine and codeine must therefore be fixed at 7:8.[22]
(b) Thebaine
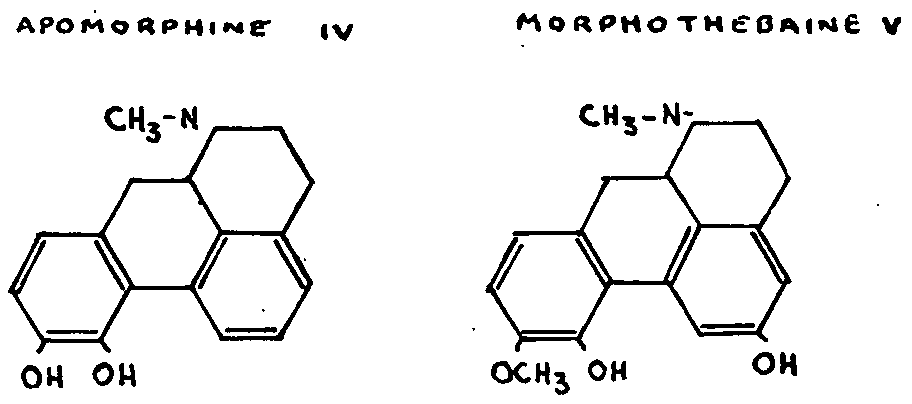
The formula for thebaine shows that it contains one double bond more than codeine. One of these double bonds is situated at 6:7, since the formation of dihydrothebainone IX,[7] referred to in paragraph 3 above, is undoubtedly a case of hydrogenolysis similar to the cases we have just mentioned. Moreover, the acid hydrolysis of thebaine and dihydrothebaine,[7] which give codeinone and dihydrocodeinone respectively-a normal reaction of enolic ethers-makes the existence of an enol extremely likely.
The conjugation of the second ethlyenic double bond of thebaine with the first has been proved by, inter alia, the recent research of Sandermann and Schöpf[23] (1938), who showed that thebaine lends itself to the Diels-Alder condensations with maleic anhydride and quinones. The addition of maleic anhydride, for example, gives a compound XI of which the methoxy group no longer reacts to acids.
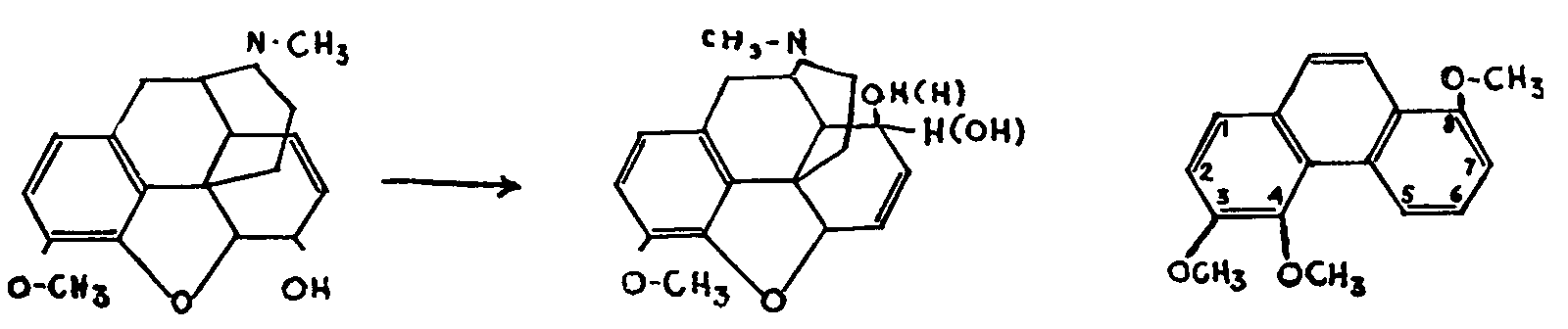
ALLO-CODEINE 3,4,8-TRIMETHOXY-
ALLO-PSEUDOCODEINE PHENANTHRENE VI
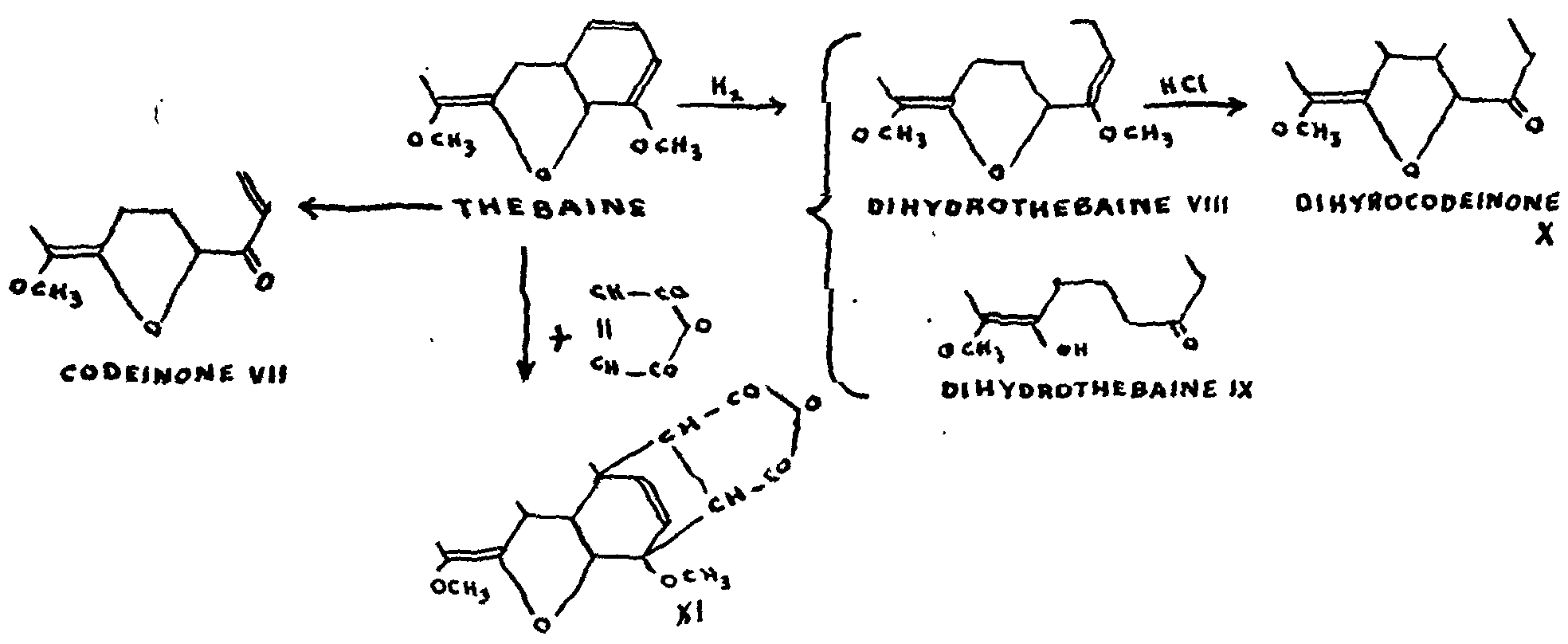
The following formulae illustrate these thebaine reactions:
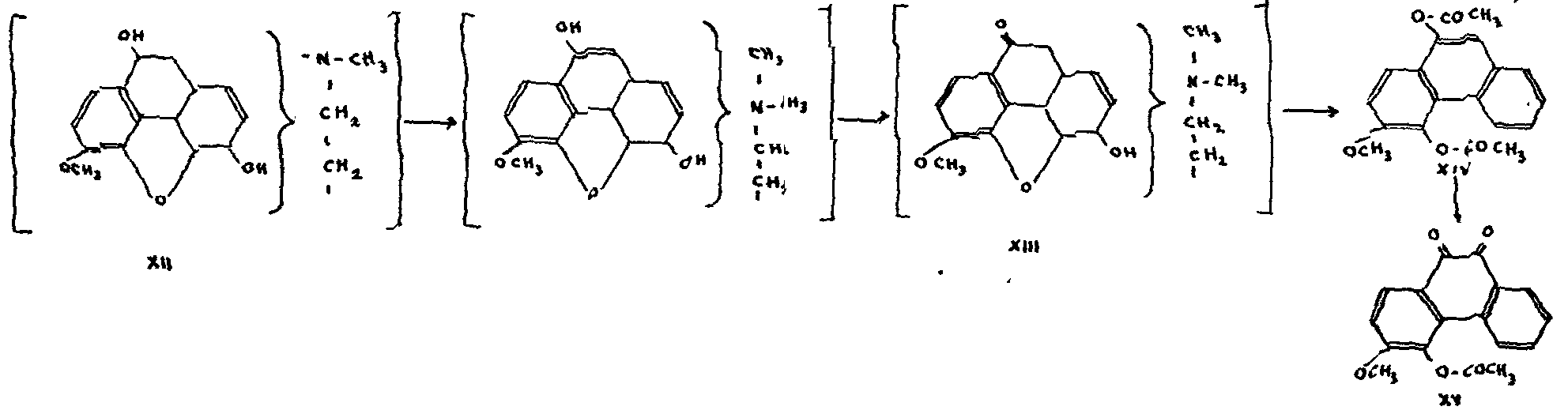
10. The nitrogen atom is linked to C-9
This deduction is supported by Knorr's work (1906) on hydroxycodeine XII. Prepared by the controlled oxidation of codeine,[24] hydroxycodeine results from the attachment of one hydroxyl to C-10, since exhaustive methylation[25] gives a ketone XIII which, when treated with acetic anhydride, yields a methoxy-diacetoxyphen-anthrene XIV oxidizable into methoxyacetoxy-phenan-threnequinone XV.
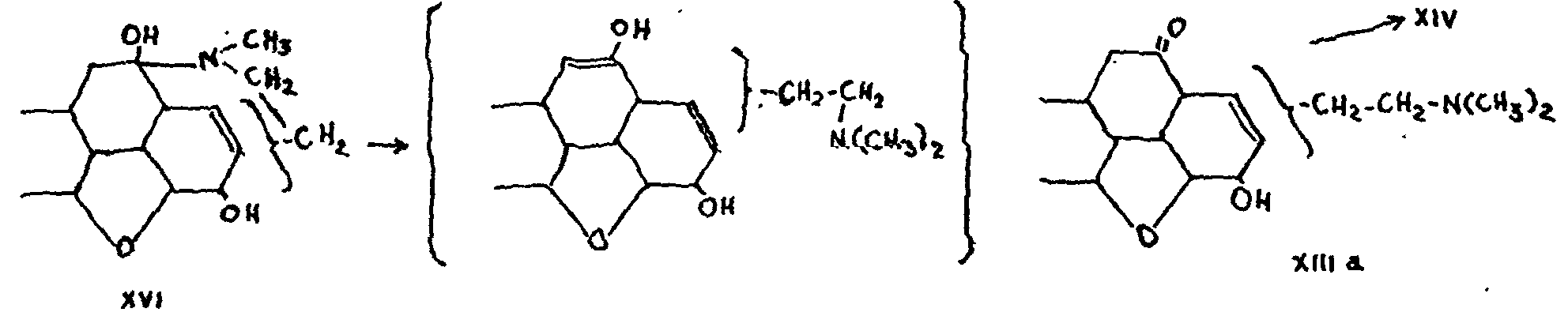
The loss of one of the acetoxy groups in the process of oxidation can be explained only if the alcoholic hydroxyl of the hydroxycodeine was located at 9 or 10. Since, moreover, compound XIII is ketonic, the hy-droxyl must necessarily be attached to the same carbon atom as the nitrogen, or to an adjacent carbon atom. Knorr adopted the former hypothesis and explained the series of reactions in the following way:
Although it has recently been established[26] that Knorr's formula XVI is unlikely to be correct with regard to the position of the extra hydroxyl of the hydroxycodeinone, his remaining deductions still hold good. It is clear, in particular, that the nitrogen is attached to the phenanthrene nucleus at C-9, while the position of hydroxyl must be at 10. An inversion of these positions is just possible, with the nitrogen at 10 and the hydroxyl at 9, but it would then be difficult to understand why codeine should oxidize into hydroxycodeine.
On the basis of the general conclusions to be drawn from the whole of the foregoing discussion, the formula for morphine may be represented as follows:
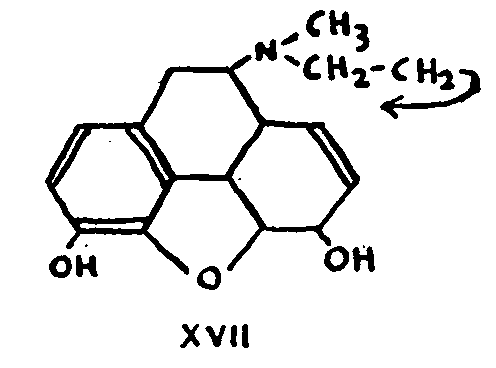
11. Nature of the nitrogen nucleus
Some of the degradation reactions referred to above separate the nitrogen of the alkaloid from the phenan- threne nucleus in the form of dimethylethanolamine.[27] As iodomethylation preceded degradation proper, it must be concluded that the -N(CH 3)-CH 2-CH 2 chain was originally linked to the phenanthrene nucleus.
At first it was claimed[28] that this chain belonged to an oxazine XVIII, "morpholine", whence the name "morpholine''[29] for the nucleus XIX from which the morphine seemed to be derived. It was later recognized that the oxygen of ethanolamine in reality formed no part of the original molecule and was the result of a secondary hydration of a vinylamine.
In 1902,[30] after explaining the structure of morphenol II, Pschorr proposed formula XX, which related morphine to apomorphine IV.
This formula in turn had to be abandoned as the result of the work on pseudocodeine (see 9 (α)), the alcoholic hydroxyl of which is in position 8-a position which would not be free if the heterocyclic nitrogen chain were attached to it.

The only remaining possibilities were that the nitrogen chain CH 2-CH 2-N(CH 3) was attached at position 5, 7, 13 or 14.
Formula XXI, Which was proposed by Knorr and Horlein in 1907,[31] was accepted in principle for almost twenty years, discussion being confined to the existence or the position of the double bond.
Attachment at 7 could not be defended after it was proved that dihydrocodeinone X, prepared by reduction of codeinone VII or by acid treatment of dihydrothebaine VIII, possessed a CO-CH 2 group.
Nor can position 14 be accepted since, if it were, there would be no place for the conjugated double bonds of thebaine.
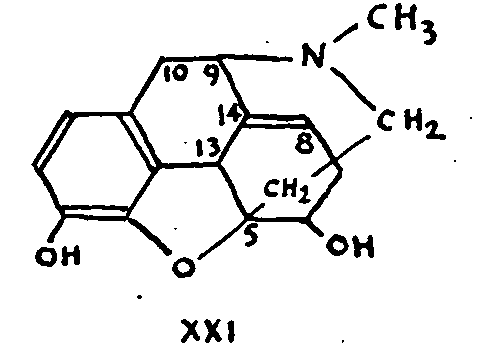
The structure formulated in XXI was criticized in 1925 by Gulland and Robinson as providing no explanation for the ease with which C-15 and C-16 separate in the reactions producing aromatic compounds (see (8)).[32] This elimination can only take place (and then it does so effectively) if there is a possibility of transforming the original system into a more stable phenanthrene system. Where this transformation is not possible, in the case of derivatives without a double bond, for example, or in the case of 14-hydroxycodeine, C-15 and C-16 do not separate. Hence their position must be such that aromatization is impossible without their elimination. Position 5 does not satisfy this condition.
Gulland and Robinson then proposed formula XXII for morphine, which is the most widely accepted today.
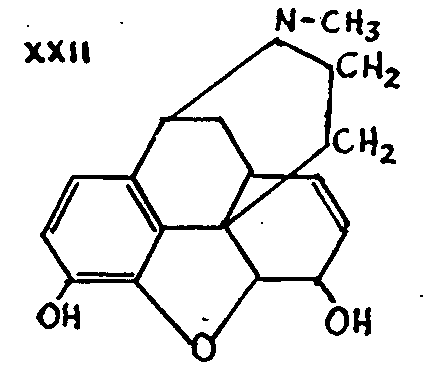
The Gulland-Robinson formula XXII received considerable support from Schöpf's work in 1927[33] on dihydrocodeinone-oxime X. Subjected to a Beckmann rearrangement (second type) the oxime is converted into aldehyde XXIII. Knorr's formula XXI should have given the ketone XXIV.
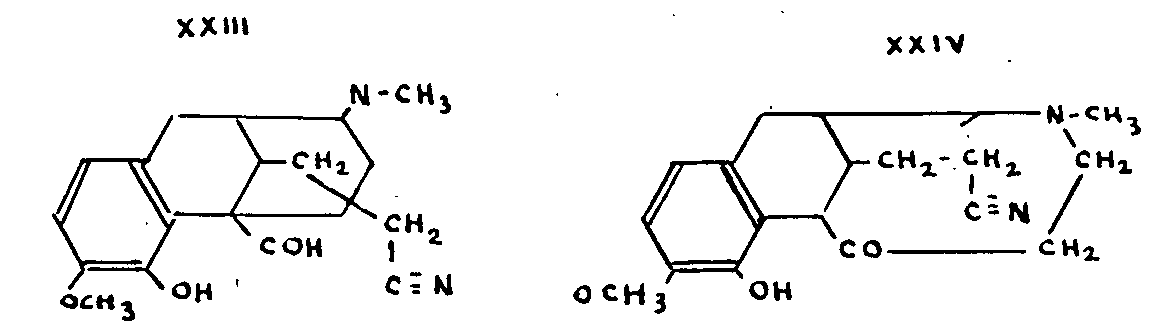
This research work, together with Schöpf's discussion of the stereochemical aspect of the Gulland-Robinson formula, has established that formula more firmly than ever.
This formula, moreover, lends itself particularly well to the explanation of a large number of complex reactions of morphine and its derivatives which cannot be dealt with here.
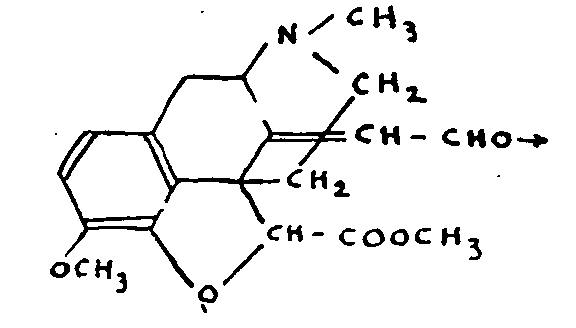
We consider it more expedient to emphasize and discuss some of the observations which do not appear wholly to agree with the formula and which make it impossible to regard the problem of the constitution of morphine as definitively solved.
(a) Pseudomorphine
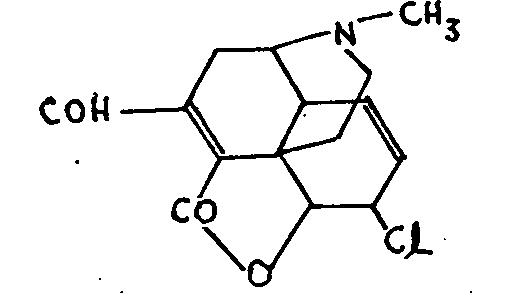
Pseudomorphine, C 34H 36O 6N 2, is easily obtained by a number of methods, including the oxidation in air of a solution of alkaline morphinate, and results from the coupling of two morphine molecules. On the analogy of the bimolecular products of the oxidation of naphthols, it has been suggested that pseudomorphine is a result of the union of two morphine molecules at C-1 or C-2. A symmetrical formula, however, cannot take into account the fact that one only of the resulting compound's four hydroxyls possesses the properties of a phenol (loss of the phenolic function as a result of mono-methylation).
Again, the two nitrogen atoms are not identical in their properties as is shown first of all by the impossibility of adding CH3I. Moreover, oxidation of morphine-methiodide gives the molecule C 17H 18O 3N(CH 3) I-C 17H 18O 3N(CH 3) OH, which is converted to diiodmethylate by hydriodic acid. Only one, however, of these two iodine atoms is easily eliminated by bases, the other being much more resistant (Vongerichten, Polstorff).[34] In 1934, Small[35] described the synthesis of similar products by the oxidation of morphine isomers (corresponding to the codeine isomers referred to in (9)). Of these the product derived from isomorphine shows the same peculiarities as pseudomorphine; its acetylated deriva-tive, however, forms an iodomethylate in the normal way.
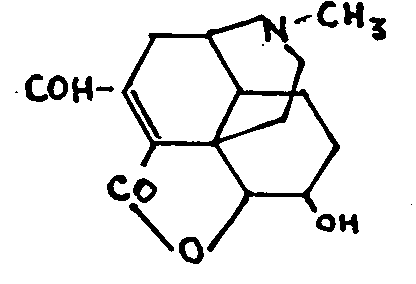
(b) Ozonization
While thebaine behaves normally with ozone to give thebaizone (Pschorr[36] 1907, Wieland, Small[37] 1928), morphine and its derivatives, whether or not containing a double bond in the carbon nucleus, suffer attack on their aromatic nucleus. Thus morphine and codeine give the same aldehyde-codinal-resulting from the loss of two carbon atoms in the case of morphine, and of two carbon atoms and one methoxy group in the case of codeine.[38]
α-Chlorocodide also suffers attack on its aromatic nucleus and gives α-chlorocodinal (Wieland, Small, Speyer, Roell):
dihydromorphine similarly gives dihydrocodinal,[39] which may also result from the action of ozone on dihydrocodeine, by way of ozodihydrocodeine, [40]
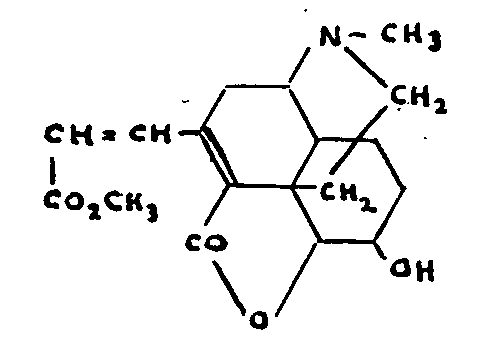
It is difficult to understand why the ethylenic double bonds of morphine and codeine do not react to ozone although behaving quite normally with other agents.
(c) Behaviour of tetrahydrodesoxycodeine
This-base is obtained by the reduction of a number of codeine derivatives. Although several of them are true phenols, tetrahydrodesoxycodeine itself is insoluble in the bases and cannot be acetylated, methylated or ben-zoylated by the usual methods.[41] This insolubility in the bases persists even after exhaustive methylation.
(d) Action of Grignard reagents on certain codeine derivatives having a double bond at 6:7
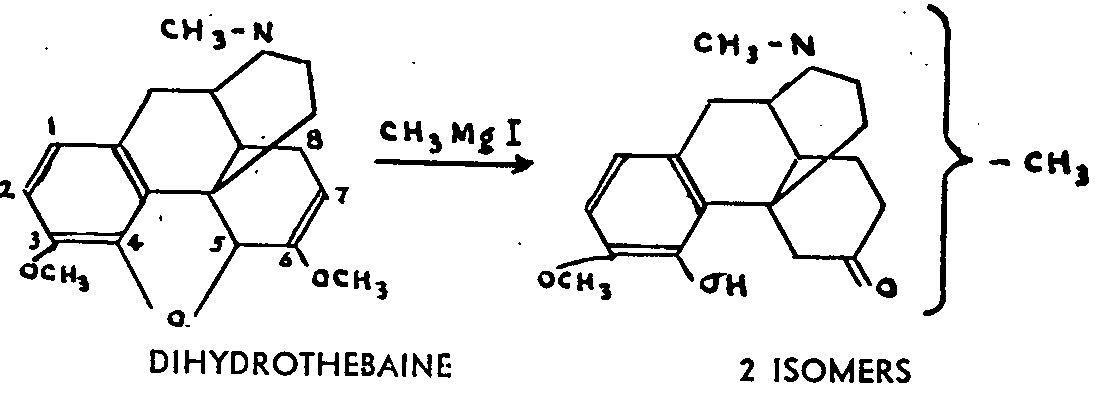
Although the points referred to in ( a), ( b), and ( c) represent surprising anomalies of behaviour,, the objections to which they give rise are not considerable enough to challenge the Gulland-Robinson formula. The point we are now' about to discuss has not yet received any final explanation and has once more thrown doubt on that formula.
The action of Grignard reagents on morphine alkaloids was studied for the first time by Freund[42] in the case of thebaine at the beginning of this century and, more recently, by Small and his collaborators. The be-haviour of thebaine, which is particularly complicated ,will be considered last of all.
Generally speaking the ketones of the codeine series do not react to Grignard reagents and their possible enolisation has been suggested as an explanation of this anomaly.[43] Codeinone, for example, does not react even at 170 degrees, at which temperature a decomposition takes place. Dihydrocodeinone and dihydropseudo-codeinone are also inert. Dihydrocodeinone, on the other hand, has been observed to react normally with methyl-lithium; it has also been noted that compounds possessing a double bond at 6:7 react abnormally to organic -magnesium compounds, whether or not their molecule contains a ketonic carbonyl group.
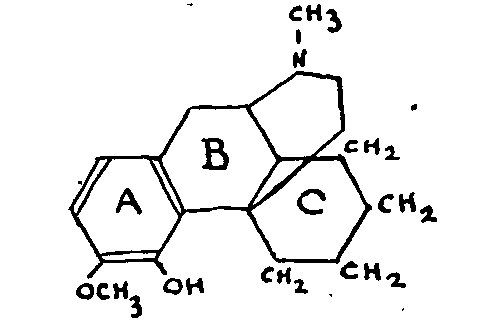
In accepting the results for thebaine which are given later, the results of the study of the following compounds have been published:
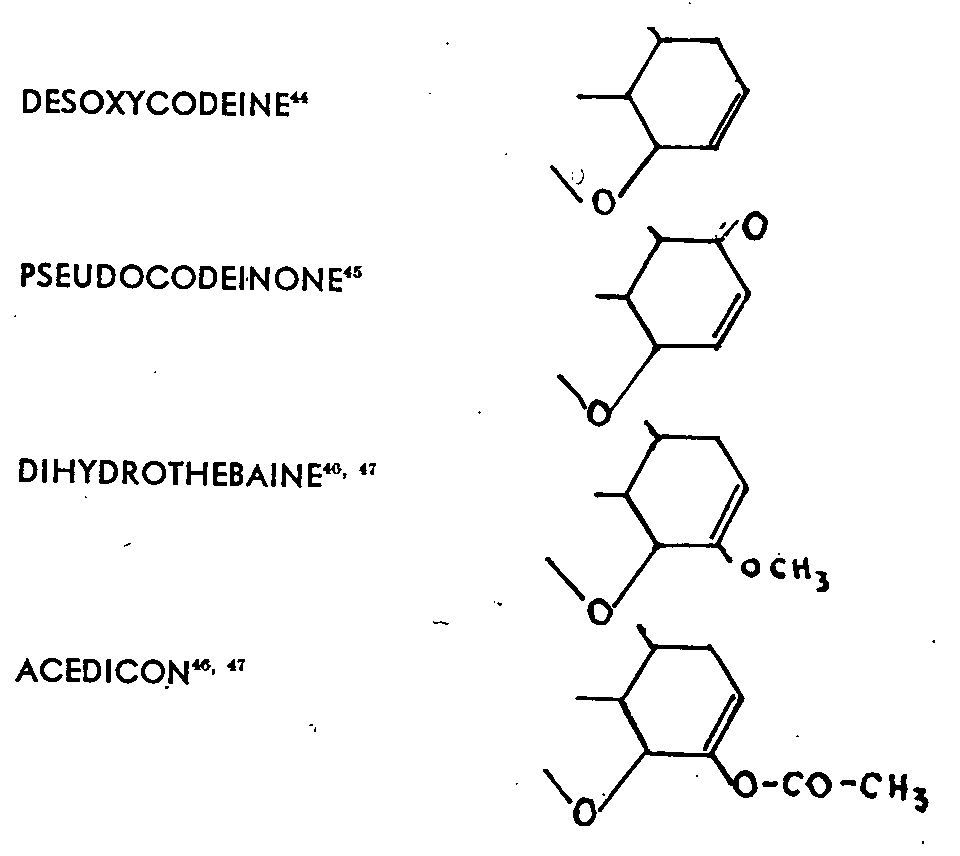
The latter two alkaloids have the peculiarity of react-ing more easily in a benzene solution than in an ether solution; whatever the magnesium reagent used, addition occurs, with saponification of the enol group' and formation of two isomers. Small, for example, showed that dihydrothebaine adds methyl magnesium iodide with fissure of the oxygen bridge and formation of a phenol group, while the enol ether hydrolysis gives a ketonic carbonyl. The reaction could, as a first approximation, be written out as follows, with the emphasis that the ketone obtained is in two isomeric forms:
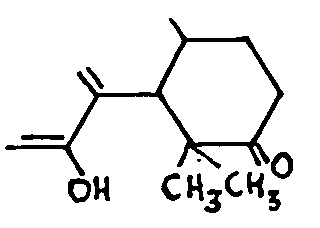
The same two isomers may be obtained from dihydro-codeinone enol acetate by means of methyl magnesium iodide. Small regarded these ketonic isomers as the re-suit of adding Grignard's reagent to the allyl system comprising the epoxyde and the double bond,6:7. It would also be possible to have a normal addition to this double bond, so that the isomers could be "either the stereoisomers of the methylated ketones ( a) at 7 or ( b) at 5; or ( c) the isomers of position, one of which would be ketone methylated at 5 and the other at 7. As we shall see, there are serious objections to each of these three hypotheses.
It should be noted that the ketonic function created by the Grignard reaction has the usual properties of the ketonic carbonyls (reduction,oximation, etc.).The iso-merism, however, survives, the bromination of the two isomers, followed by the action of a base. This treat-ment re-establishes the oxygen bridge and produces two methyldihydrocodeinones, one of which, dimethylated with hydrobromic acid gives metopon, the pharmaco-dynamic properties of which have aroused considerable interest:
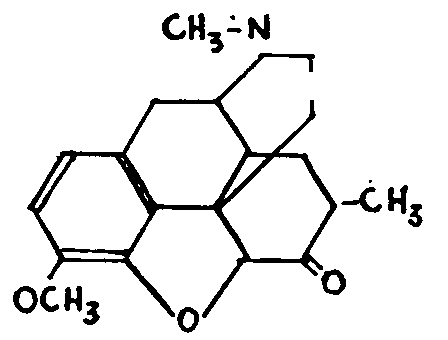
POSSIBLE FORMULA OF METOPON, AFTER SMALL
The particular objections raised by each of the three hypotheses ( a), ( b), and ( c), put forward above to explain the isomerism, may be summed up as follows:
Hypothesis ( a), that methylation takes place at 7 with formation of two diastereoisomers, must be rejected for two reasons: the two methyldihydrocodeinones react with ethyl oxalate, indicating the presence of the group -CO-OH 2- and not of the group -CO-CH-R. (The be-haviour of metopon confirms this conclusion.)
The two methyldihydrocodeinones form different enolic acetates, of which the double bond must be at 6:7, since their reaction with methyl magnesium iodide gives one and the same dimethyldihydrothebainone in each case. The latter compound is no longer able to re-store its epoxyde, which could be explained if dimethy-lation at 5 were accepted.
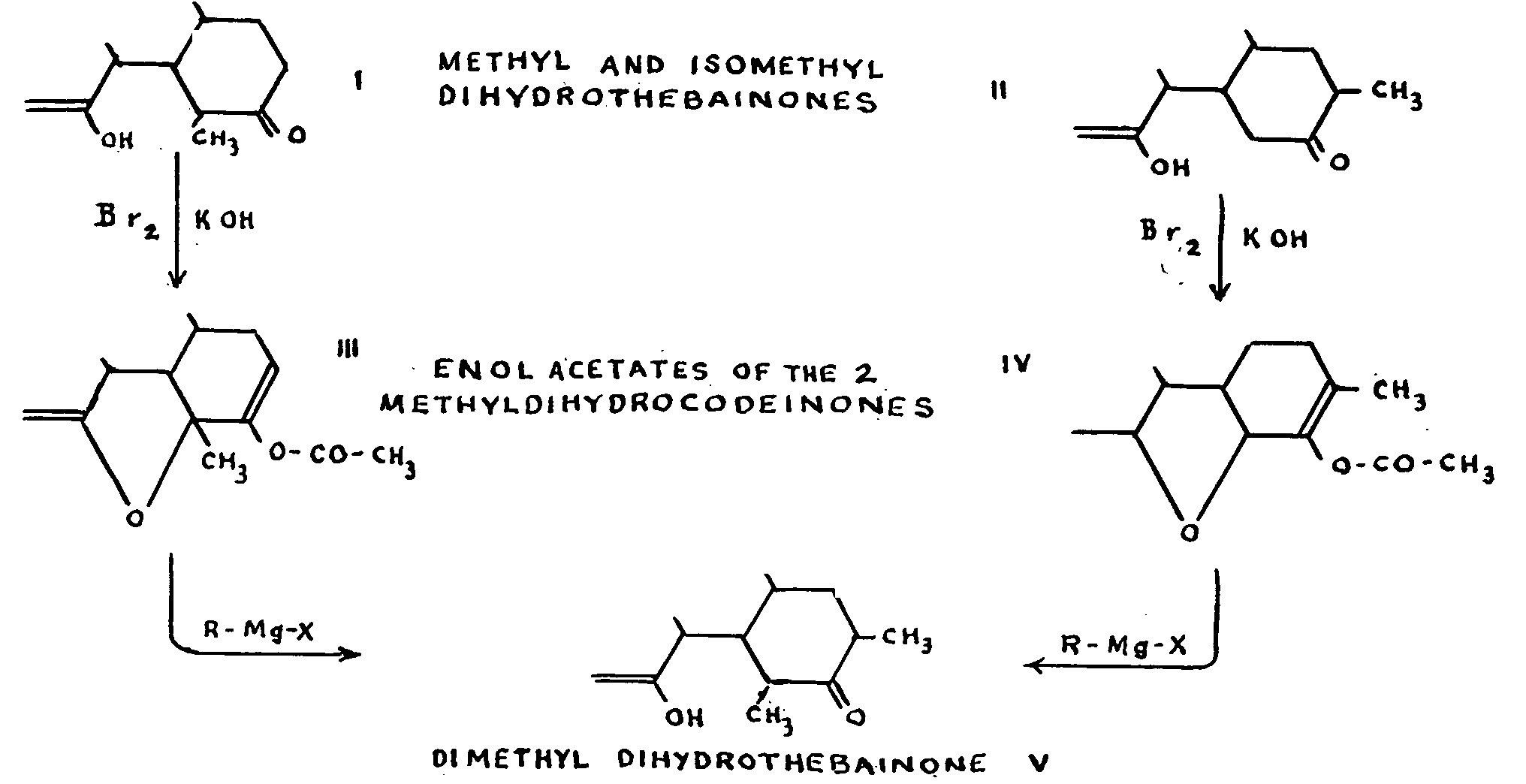
We must now consider hypothesis ( b) -that the stereoisomerism is the result of a methylated substitu-tion at 5 in the two diastereoisomers: this would imply that the two methyldihydrothebainones should, by the action of the bromine and the bases, give the same methyldihydrocodeinone, since only a single structure at 5 is compatible with the existence of an oxygen bridge. This deduction, however, is refuted by the formation of two epoxydic isomers, as stated above.
We are thus reduced to the last of the three.hypothe-ses, hypothesis ( c). This is incompatible with the ethyl oxalate reaction and with the fact that it is impossible to store the oxygen bridge in the dimethyldihydro-thebainone.
The fact that reactions I --> III can be obtained shows that if the methyl is in position 5 the structure is conducive to the formation of the bridge. This favourable structure should be transmitted to V, which should thus, contrary to experience, be able to re-establish its epoxyde.
It is possible that attempts at degradation into phenanthrene derivatives may decide which of these conflict-ing hypotheses is correct. Nothing seems as yet to have been published on the subject, but it is possible that industrial laboratory work on metopon, as yet unpub-lished, has already shed some light on these complicated questions of structure. The only recent publication which suggests that the solution of the problem is on the way is that of Small,[48] who formulates metopon as a 7-methyldihydromorphinone, but does not give his reasons.
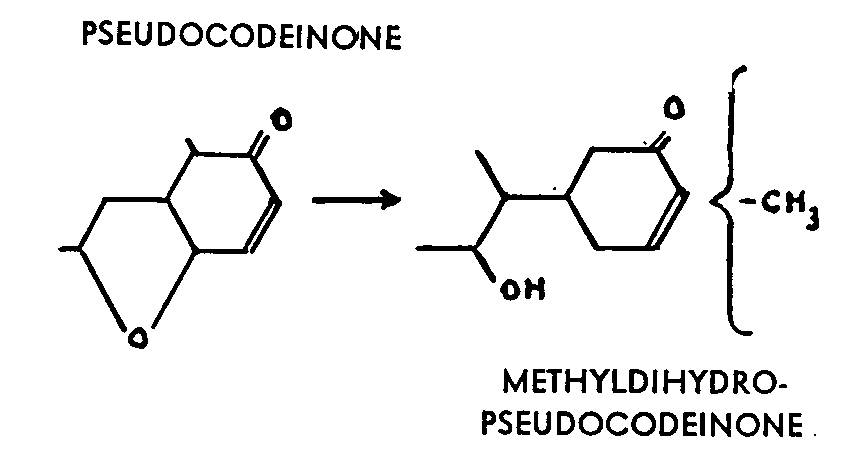
Some of the reactions between magnesium compounds and morphine derivatives which have been studied and published may be regarded as constituting the transi-tion from the cases which have been discussed above to the even more confused case of thebaine. Methyl magnesium iodide, for example, acts on pseudocodeinone to form methyldihydropseudocodeinone, which is phenolic and should, from its composition, be the result of the addition of a methyl and of a fission of the oxygen bridge.
This compound, too, shows several anomalies: it is particularly resistant to hydrogenation and forms neither an oxime nor a semi- carbazone. Its analog, derived from pseudocodeine methyl ether, cannot be reduced either. it reacts with Grignard reagents to form phenol compounds as the result of the addition of the organic radical and fission of the organic bridge. In the case of the phenyl and the ethyl, small quantities of isomers are produced, some of which resist catalytic hydrogenation.
(e) Action of Grignard reagents on thebaine
This study was begun by' Freund in the hope that it would quickly provide an explanation of the structure of thebaine and it has proved to be one of the most complicated and hermetic in the whole of organic chemistry. Not all its secrets have yet been solved, despite the substantial progress recently made.
As far back as 1905, Freund succeeded, by treating thebaine with phenyl magnesium bromide, in isolating a phenol compound, phenyldihydrothebaine, which, it is now believed, is the result of the addition of the phenyl with fission of the oxygen bridge and consists of two isomers. This molecule could be represented provisionally by one of the following formulae:
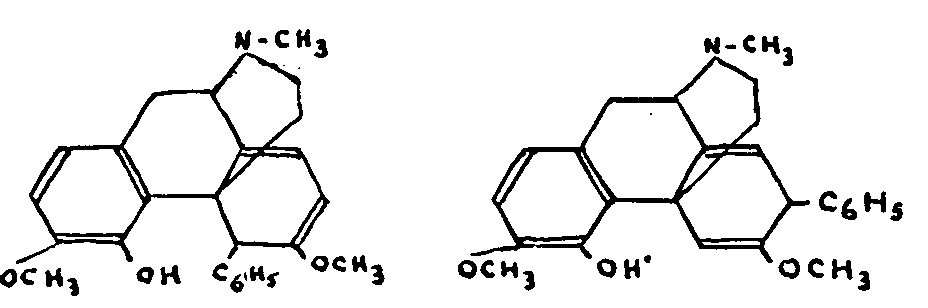
This compound shows many anomalies:
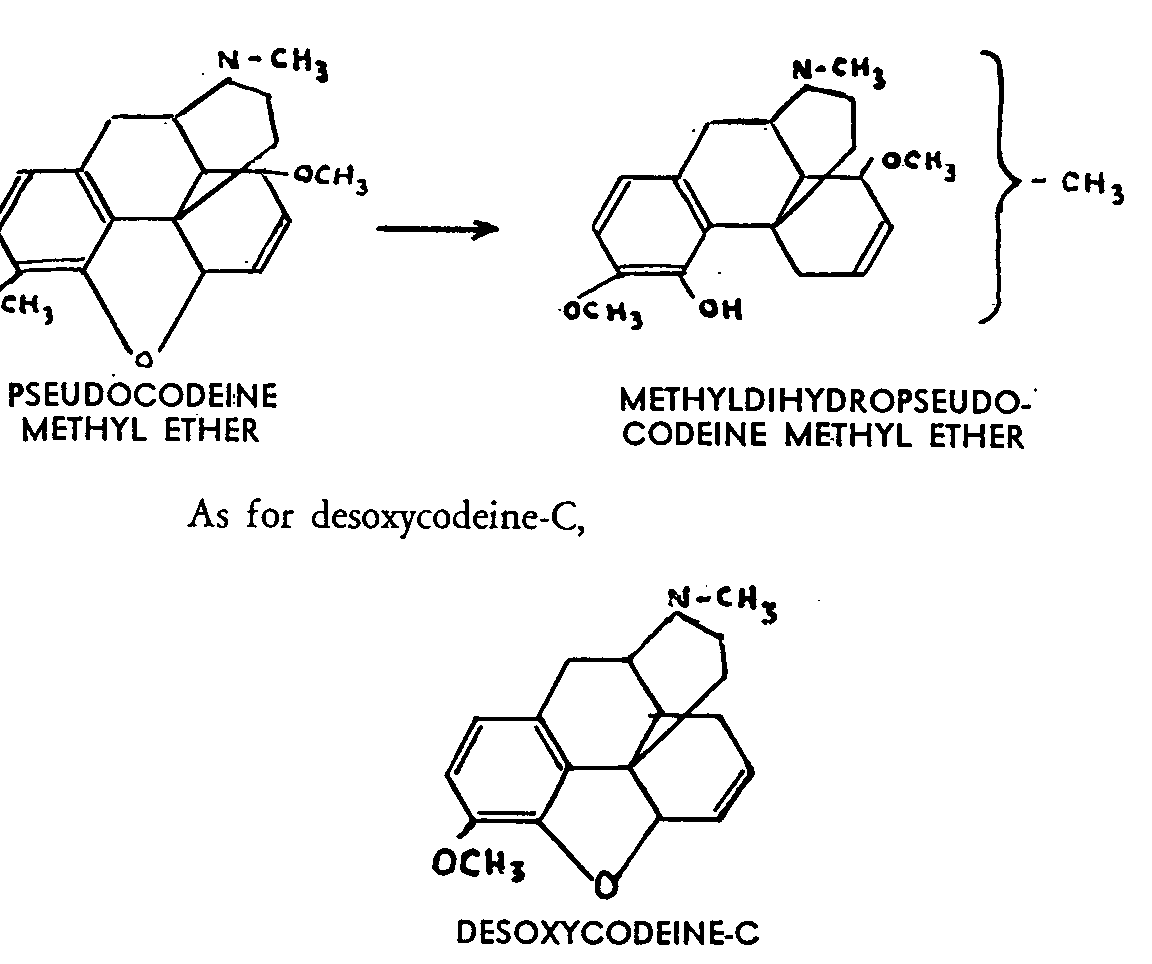
The double linkages are so resistant to hydrogenation that they are unaffected by the most powerful methods; the compound is stable towards acids and bases. Boiled with acetic anhydride it gives an acetic ester, while in the same conditions thebaine is degraded into acetylthebaol. Its methyl ether does not react with ozone. One of its methoxyl groups has lost its enolic character, since it is no longer affected by boiling with hydrochloric acid. Its complete demethoxylation is possible with hydrochloric acid under pressure, but the resulting products is not ketonic, behaves like a triphenol and through the action of diazomethane, changes into phenyldihydrothebaine methyl ether. Aromatization of the carbon nucleus would account for all these characteristics, but the hydrogen content of the product would be incompatible with this hypothesis unless there have been considerable changes in the carbon skeleton. Lastly, Hofmann degradation of the phenylated (or methylated) isomers yields two isomeric methines through elimination of the nitrogen, although, curiously enough, the two carbon atoms linking the nitrogen to the original molecule are retained.
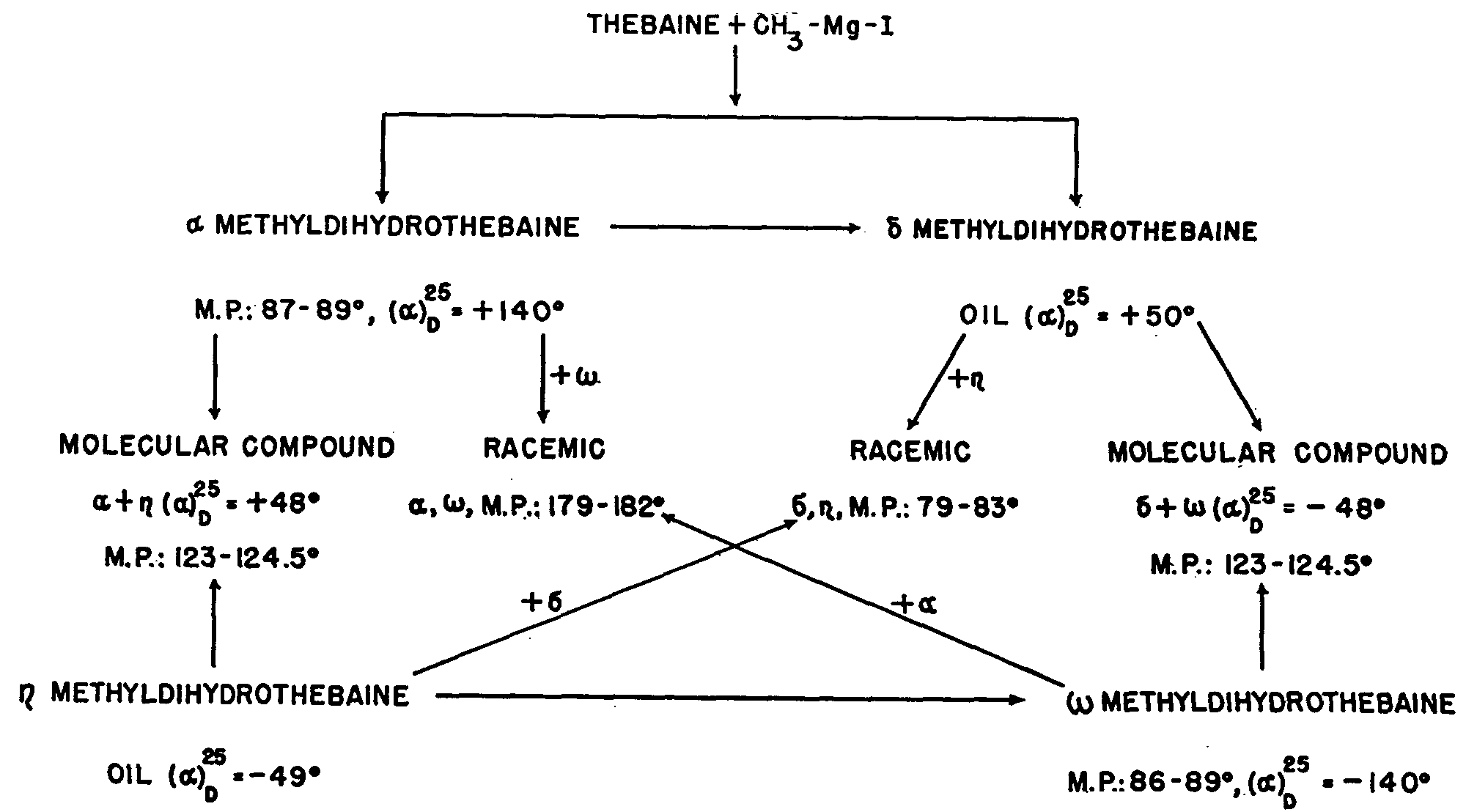
Small and Fry tried to find an explanation of this abnormal behaviour by studying the reaction between thebaine and methyl magnesium iodide. Two isomeric methyldihydrothebaines, α and δ were isolated, both phenolic, with the same resistance to hydrogenation and acid action as the corresponding phenylated derivative of Freund: each of these two isomers may be isomerized in turn by heat, so that it is possible to get, finally, four isomeric methyldihydrothebaines, α, π, δ, ω. The methods of obtaining these isomers and their interrelations are tabulated below:
The four isomers can be seen to group themselves into two pairs of optical antipodes, implying two centres of asymmetry, and two only, in the molecule.
In 1947, Small, Sargent and Bradley published the results of studies originally designed to verify Freund's work but actually clarifying, expanding and supplementing it. The phenylated series follows the behaviour described above for the methylated series in every respect. It is possible, in particular, to isolate four isomeric phenyldihydrothebaines which are arranged in two groups of optical antipodes, and of which the Hofmann degradation is also abnormal.
In the light of all the observations collected and after a very detailed discussion of the problem, Small first proposed a series of formulae which accurately reflect the interrelations between the various thebaine derivatives but are unable to account for their properties or their numerous anomalies.He was then led to question the validity of the Gulland-Robinson formula for morphine.
In a paper read before the Royal Society in 1947 49 Robinson suggested an unexpected and wholly original solution which would leave his formula for thebaine unaffected. Unfortunately, however, the awaited detailed publication has not yet been made. Robinson stated the principle that the carbon nucleus of the compounds resulting from the action of an organic magnesium compound on thebaine must be aromatic; this accounts for their behaviour and all their observed anomalies, such as their resistance to hydrogenation, the acquired resisance of the previously labile methoxide and the strongly phenolic character of the product of demethylation.
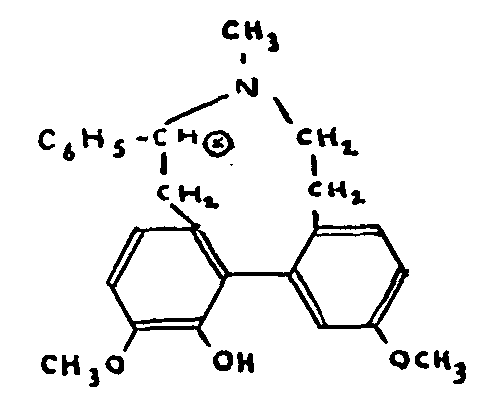
The products of two Hofmann degradations, in which the nitrogen is eliminated, retain their optical activity. Since it is clear, however, that none of the 24 carbon atoms remaining in the denitrified molecules (18 for the 3 nuclei, 4 to support the two double bonds and 2 belonging to the methoxy group) can possibly be asymmetrical and thus account for the continued optical activity, Robinson evolved the hypothesis of a molecular asymmetry based on a diphenyl structure of which the two nuclei are of different planes.
The second centre of asymmetry which must be present in the phenyldihydrothebaines would be an asymmetrical carbon atom belonging to a nitrogen heterocycle of nine atoms and replaced by the phenyl radical.
Robinson therefore proposed the following formula: the formation of the phenyldihydrothebaines from the- baine would follow a plausible sequence developed by Robinson. These conclusions can, of course, apply to the various products of the action of organic magnesium compounds on thebaine.
The Robinson hypothesis and formula not only provide a satisfactory explanation of what was previously regarded as anomalies, but are also in harmony with the studies made of the ultra-violet absorption of these various compounds.
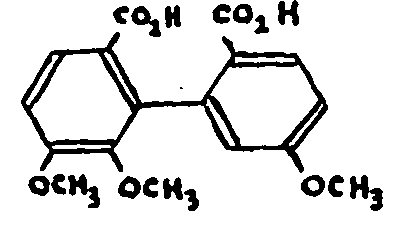
It should further be noted that Robinson claims to have obtained the diphenyl acid when oxidizing the products of exhaustive methylation.
It is clear from the foregoing that the various transformations of the morphine series can, in general, be explained by using the Gulland-Robinson formula; nevertheless there remain a number of difficulties, the most important of which arise out of the study of the action of Grignard reagents.
The best proof of the validity of the Gulland-Robinson formula would be provided by a total synthesis, not of morphine or codeine, but of one of their derivatives whose method of formation is not disputed.No synthesis of this kind has yet been completed, although there are certain reliable indications that it may be expected in the near future.
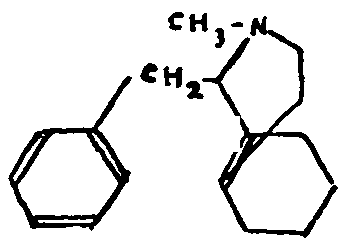
Grewe's recent work is the most promising. After a series of studies of the partially hydrogenated phenanthrene derivatives, Grewe has succeeded by a highly original method of synthesis in isolating N-methylmorphinane, by cyclizing 1-benzyl-N-methyl-octohydroisoquinoline, under the influence of phosphoric acid.
As we see, N-methyl-morphinane, the constitution of which has been established by Grewe, is closely related to the natural alkaloids. Tetrahydrodesoxycodeine would thus be 3-methoxy-4-hydroxy-N-methyl-morphinane.
The Hofmann-La Roche laboratories have taken a further step towards the solution of the problem by synthesizing 3-hydroxy-N-methyl-morphinane in two different ways. The sole remaining difference between the methyl ether of this compound and tetrahydrodesoxycodeine is the absence of the phenolic hydroxyl at C-4. Hence, all that remains is to eliminate this hydroxyl in the natural alkaloid or, conversely, to synthesize the molecule containing it. A direct comparison between the natural and synthetic products would be decisive.
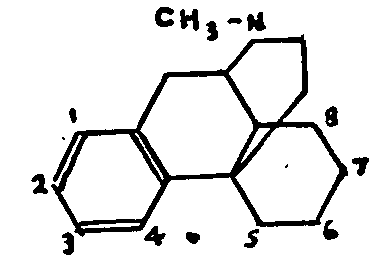
It should be noted that racemic 3-hydroxy-N-methyl-morphinane (its separation has not yet been accomplished) is a very powerful analgesic, several times as strong as morphine.
This compound therefore deserves very special study not only because of its synthetic origin, but also for its pharmaco-dynamic properties. This study will be made in the next article which will deal with the chemistry of synthetic analgesics.
Editors' note.
At the time of going to press, we receive from the author the following information: "A highly important work has just been published in Germany by Grewe (R. Grewe, Alb, Mondon, Elisabeth Nolte: Annalen,vol. 564, P. 161) in which this author describes the total synthesis of racemic tetrahydrodesoxycodeine followed by its double decomposition. As isomer, as well as the synthetic racemic, have been identified with the corresponding compounds produced by the reduction of codeine, any remaining doubts as to the Gulland and Robinson formulae are now finally removed".
1Trommsdorf’s Journal der Pharmazie, 13 (I) 234 (1805); 14 (I) 47 (1806); 20(I) 99 (1811).
2Derosne: Ann. Chim., 45, 257 (1803); 92, 225 (1814).
4Consult: L. F. Small, Chemistry of the opium alkaloids, Washington D. C., U. S. Government Printing Office, 1932.
5Dobbie, Lauder: J. Amer.Chem. Soc., 99, 34 (1911). Van Duin, Robinson, Smith: Ibid.,903 (1926). Small: J. Org. Chem,. 12, 359 (1947).
6Junusov, Konovalova, Orekhov: Ber., 68, 2158 (1935); Zhourn. Obshchei Khim., 10, 641 (1940). Konovalova, Kiselev: Zhourn. Obshchei. Khim., 18, 855 (1948).
7Freund, Speyer, Gutmann: Ber. 53, 2250 (1920); Skita. Nord. Reichert, Stukart: Ber., 54, 1560 (1921).
8Schöpf, Winterhalder: Ann., 452, 211 (1927).
9Knorr: Ber., 39, 1409 (1906).
10Oldenberg: Ber., 44, 1829 (1911).
11Very numerous works since Grimaux, Comptes Rendus, 93, 591 (1881) and Hesse, Ann., 222, 203 (1883).
12Vongerichten: Ann. 210, 396 (1881); Ber., 31,51 (1898); Ber., 34, 1162 (1901).
13Very numerous publications since Fisher, Vongerichten: Ber., 19, 792 (1886); consult Small ( op. cit.) pp. 281, et seq.
14Vongerichten: Ber., 30, 2439 (1897); Ber. 33, 3521 (1900).
15Freund, G?bel: Ber., 28, 941 (1895).
16Knorr: Ber., 36, 3074 (1903).
17Pschorr, Sumuleanu: Ber., 33, 1810 (1900).
18Pschorr, Seydel, Stöhrer: Ber., 35, 4400 (1902).
19Späth, Hromatka: Ber., 62, 325 (1925).
20Very important bibliography in Small ( op. cit.) pp. 211, et seq.
21Gulland, Robinson: J. Chem. Soc. [London], 23, 980 (1923).
22Speyer, Wieters: Ber., 54, 2647 (1921). Speyer, Krauss: Ann., 432, 233 (1923). Lutz, Small: J. Amer. Chem. Soc., 54, 4715 (1932). Small and collaborators in later publications.
23Sandermann: Ber., 71, 648 (1938). Schöpf, .V. Gottberg, Petri: Ann., 536, 216 (1938).
24Ach, Knorr: Ber., 36, 3067 (1903).
25Knorr, Schneider: Ber., 39, 1414 (1906). Knorr, Hörlein: Ber., 40, 2042 (1907); Ber., 39, 3252 (1906). Pschorr, Einbeck: Ber., 40, 1480 (1907).
26Holmes, Lee: J. Amer. Chem. Soc., 69, 1996 (1947).
27See, among numerous works, cited by Small ( op. cit.) pp. 285-6. Knorr: Ber., 22, 113 (1889); 27, 1144 (1894).
28Knorr: Ber., 22, 1113 (1889).
29Knorr: Ann., 301, 1 (1898); Ber., 22, 2081 (1889).
30Pschorr, Juckel, Fecht: Ber., 35, 4377 (1902).
31Knorr, Hörlein: Ber., 40, 3341 (1907).
32Gulland, Robinson: J Chem. Soc., 123, 980 (1923); Mem. Proc. Manchester Lit. Phil. Soc., 69, 79 (1925).
33Schöpf, Ann., 452, 211 (1927).
34Vongerichten: Ann., 294, 206 (1896). Polstorff: Ber., 13, 93 (1880).
35Small: J Amer. Chem. Soc., 56. 1930 (1934).
36Pschorr, Einbeck: Ber., 40, 3652 (1907).
37Wieland, Small: Ann., 467, 17 (1928).
38Speyer, Roell: Ber., 63, 539 (1930).
39Speyer: Ber., 62, 209 .(1929).
40Speyer: Popp: Ber., 59, 390 (1926).
41Small, Cohen: J Amer. Chem. Soc., 54, 802 (1932); see also Small ( op. cit.) p. 240.
42Freund: Ber., 38, 3234 (1905).
43Schneider: Dissert., Jena 1906. Lutz, Small: J Amer. Chem. Soc., 57, 2651 (1935).
44Small: J. Amer. Chem. Soc., 58, 192 (1936).
45Lutz, Small: J Amer. Chem. Soc., 57, 2651 (1935).
46Small, Fitch, Smith: J Amer. Chem. Soc., 58, 1457 (1936) :
47Small, Turnbull, Fitch: J Org. Chem. 3, 204-232 (1938).
48Small: Annals N. Y. Ac. Sc., 51, 12 (1948).
49Proc. Roy. Soc. B., 35 V - XIX (1947).


