METHODS
RESULTS
SUMMARY AND CONCLUSIONS
Literature
Erie Einar Ekstrand
Author: Nathan B. Eddy, , Hedwig Besendorf , Béla Pellmont
Pages: 23 to 42
Creation Date: 1958/01/01
Braenden, Eddy & Halbach (1955), summarizing the relationship between chemical structure and analgesic action, said "In morphine and its derivatives the methyl substituent on nitrogen seems essential because its substitution by other alkyl groups reduces or abolishes analgesic action." It is well known, of course, that substituting alkyl for methyl on the nitrogen of morphine results in a compound (nalorphine) which is antagonistic to most morphine-like effects and which retains little analgesic action as measured by various techniques in animals. Recently Winter, Orahovats & Lehman (1957) reported on analgesic activity and morphine antagonism in a large series of N-substituted derivatives of morphine. In agreement with the conclusion quoted, all alkyl groups tried reduced analgesic activity. Also N-phenacyl-, N-phenoxyethyl-, and N-benzylnormorphine had less than one-tenth the analgesic potency of morphine and N-cyclohexylethylnormorphine was one-third as effective. In striking contrast they reported that N-phenethylnor-morphine was six times more effective than morphine as an analgesic in rats.
Levorphanol [(-) 3-hydroxy-N-methylmorphinan], representing an incomplete synthesis of morphine, is morphine-like in its action with a greater analgesic potency. If allyl is substituted for methyl on the nitrogen of levorphanol, the resulting compound, levallorphan, is also a morphine antagonist which dose-wise is more effective than nalorphine. In addition we have found that the (-) (3-hydroxy-N-phenethylmorphinan has more than three times the analgesic potency of its methyl analogue. Consequently, with parallelism between morphine and morphinan derivatives established in these respects, a systematic investigation of a considerable number of N-aralkyl (and N-alkyl) derivatives of 3-hydroxy- (or 3-methoxy-) morphinan was undertaken. The present paper will describe the effect of N-aralkyl substitution only.
All compounds were tested for analgesic potency in mice by two different methods, and many were tested in rats and rabbits. Toxicity determinations were made on mice.* In one series of experiments on mice the hot-plate method was employed (Eddy & Leimbach, 1953). An ED 50 for subcutaneous administration for all active compounds and an ED 50 for oral administration wherever possible were calculated by probit analysis. In a second series of experiments on mice, radiant heat was applied to the tail. Administration of the drug was subcutaneous and ten animals per dose were used. The increase of the reaction time area (minute-seconds) was measured and the MED 50 (minimal effective dose for a 50% increase of reaction time area evaluated graphically.
1Chief, Section on Analgesics, Laboratory of Chemistry, National Institute of Arthritis and Metabolic Diseases, National Institutes of Health, Bethesda, Maryland, U.S.A.
2Pharmacologist, Hoffmann - La Roche laboratories, Basle, Switzerland.
3Chief of pharmacological laboratories, Hoffmann - La Roche, Basle, Switzerland.
*Subcutaneous and oral ED 50's in mice and subcutaneous LD 50 's were determined at Bethesda; the other data were obtained in the Hoffman-La Roche laboratories at Basle. All compounds were supplied by Drs. O. Schnider, A. Grüssner & J. Hellerbach, of the Hoffman - La Roche laboratories.
For rats, radiant heat was applied to the shaved skin of the back. Five animals were used per dose, and administration was subcutaneous. MED 50 was the dose producing a 50% increase in the average maximal post-drug reaction time. For rabbits an electrical stimulus was applied to the tooth pulp (Koll & Reffert, 1938; von Gordonoff & Ruckstuhl, 1939). At least three animals were used per dose and the drug was administered intravenously. For rabbits the MED 50 was the dose after which the threshold of electrical stimulation was increased by 50%.
Two determinations of toxicity were made. In one, compounds were administered subcutaneously in mice, five or ten animals per dose. If possible, an LD 50 was calculated by probit analysis, but in many instances the low order of toxicity and the small amount of material available permitted only the conclusion that the LD 50 was above a certain level. In the second determination substances were administered intravenously to mice, using four animals per dose, and an LD 50, an LD 10 and an LD 50 were calculated.
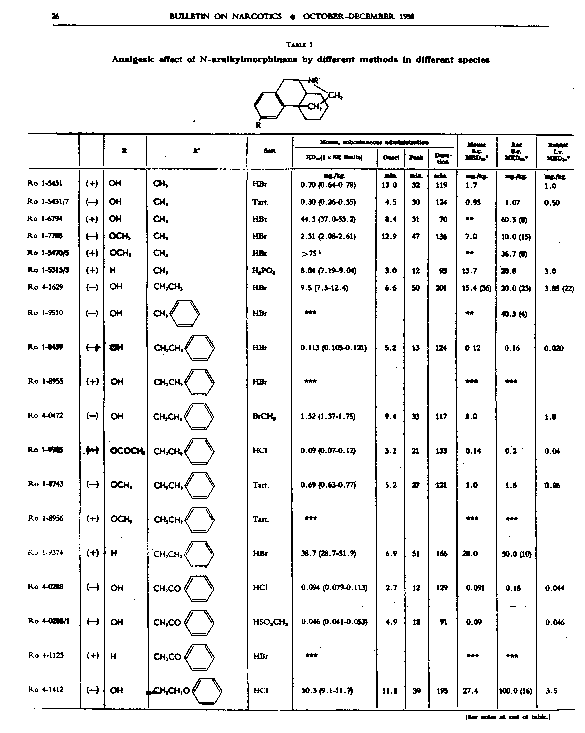
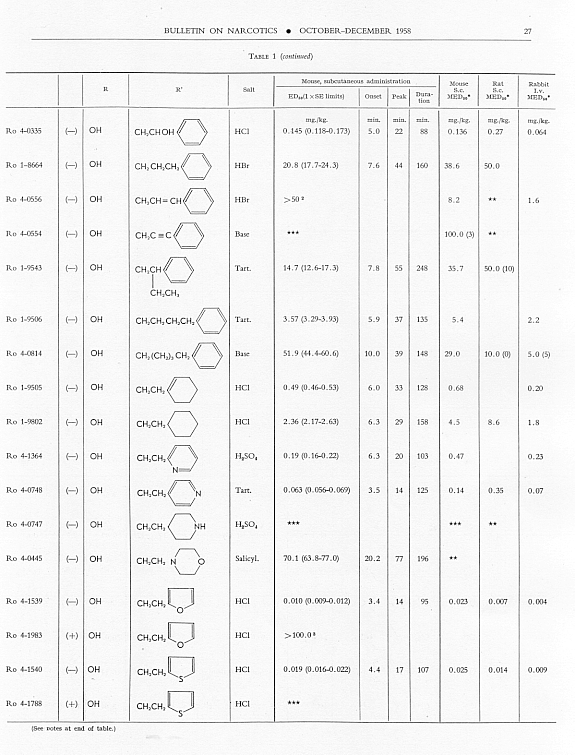
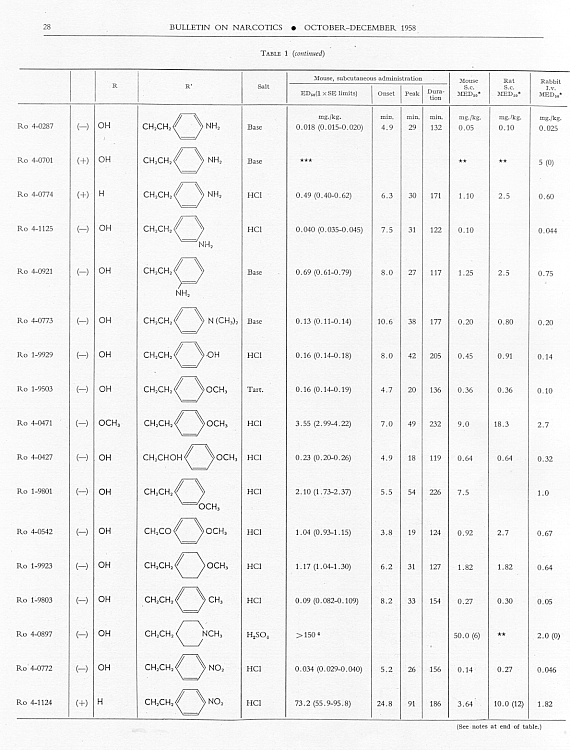
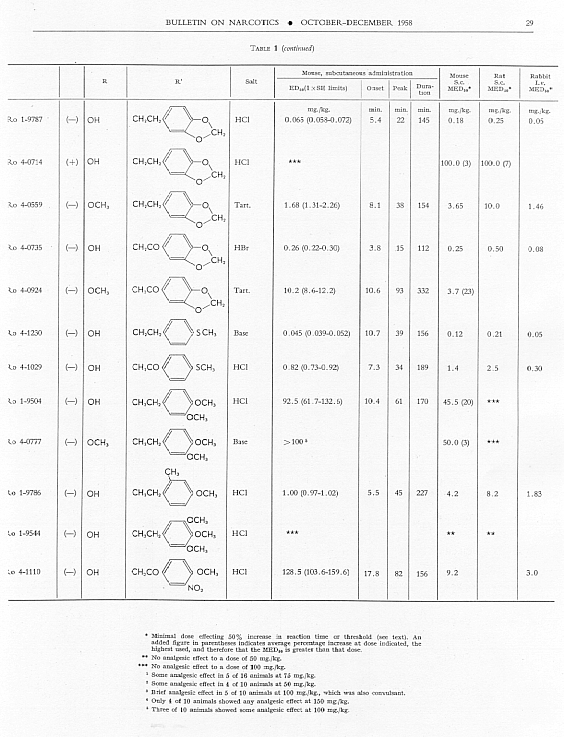
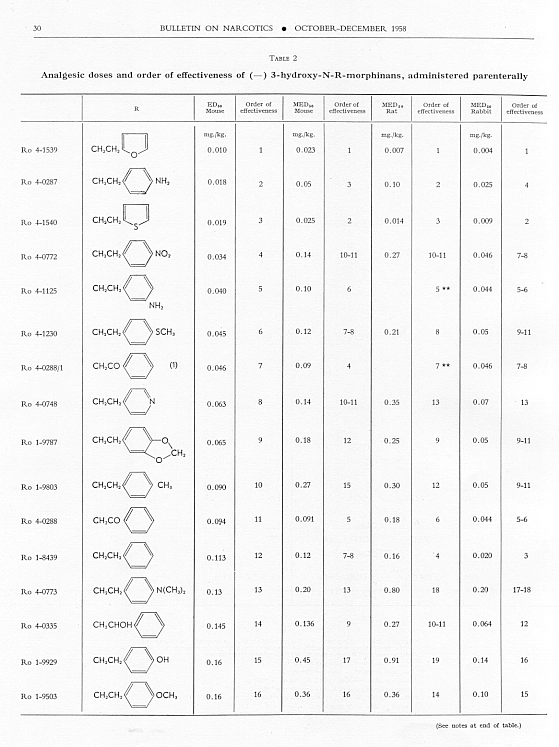
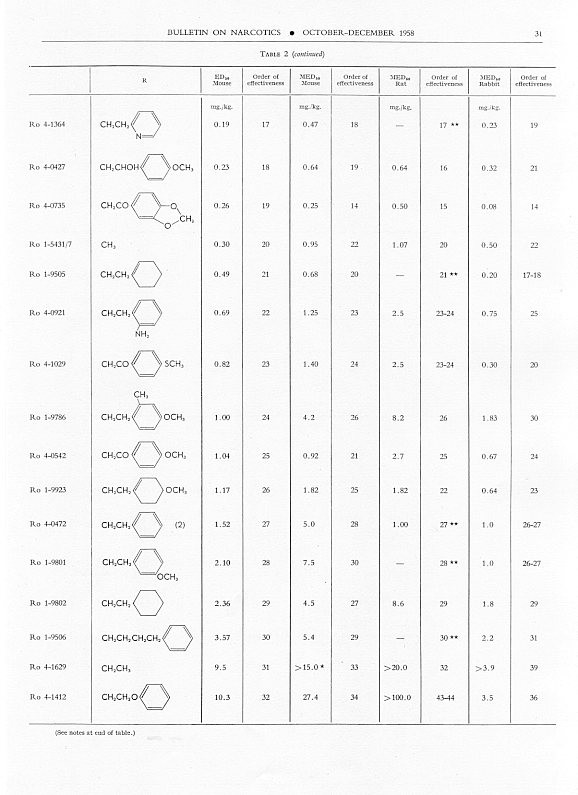
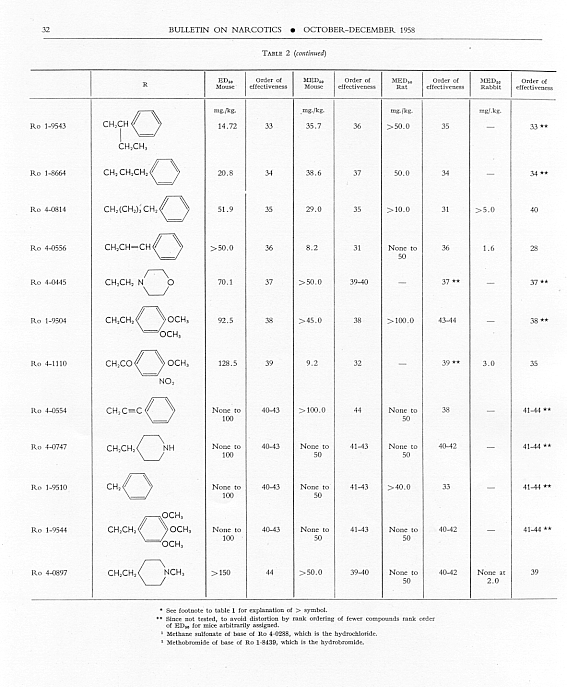
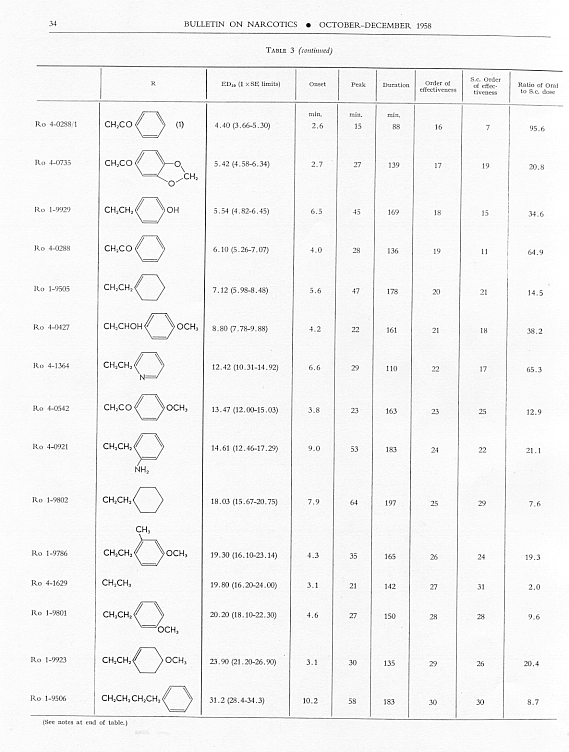
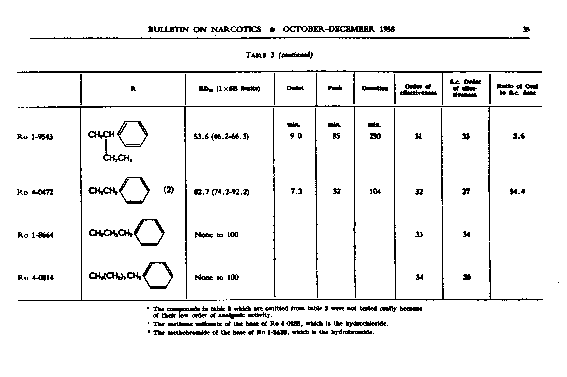
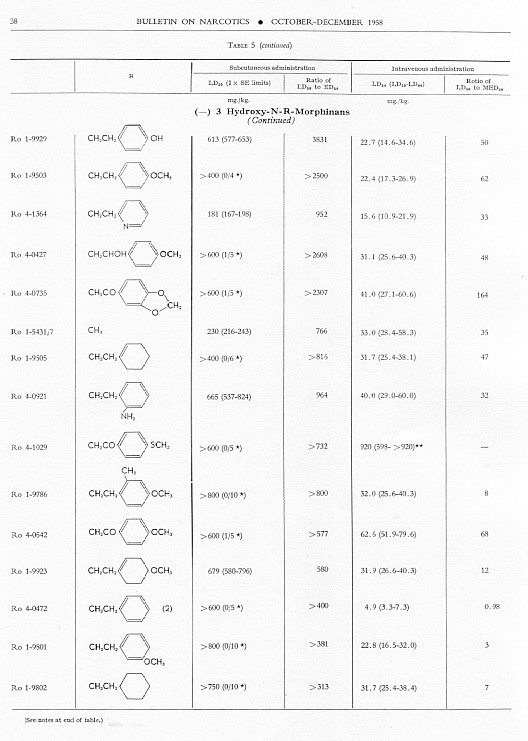
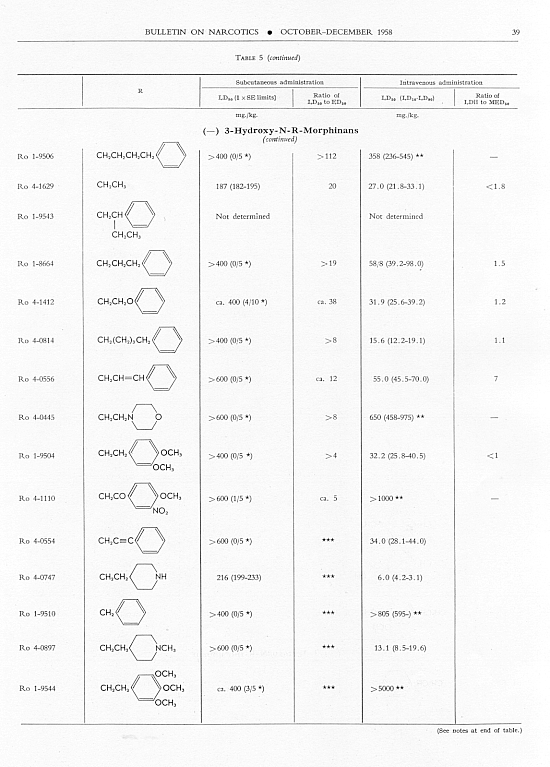
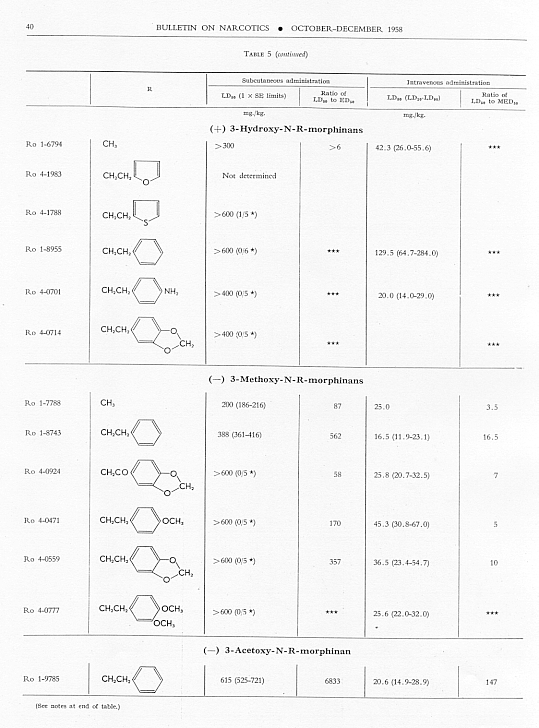
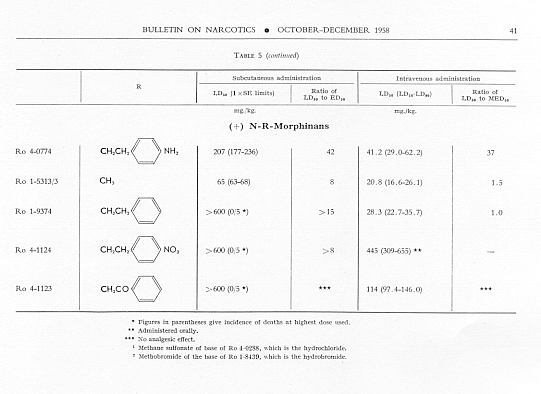
Analgesic action. The compounds studied and results obtained by parenteral administration are presented in table 1, where an attempt has been made to arrange the compounds according to chemical relationships, particularly with respect to the substituent on nitrogen. To bring out more clearly the effect of N-aralkyl substitution, the compounds have been rearranged in table 2 in order of decreasing potency, based on the ED 50 figures for mice (Bethesda observations). This table includes data for the (-) 3-hydroxymorphinans only and also presents in parallel columns the potency figures for mice (Basle observations), for rats and for rabbits. There are variations in the order of potency from one set of observations to another and from one species to another, but the over-all trend is the same for each method and each species. The trend and the significant variations will be discussed in the following paragraphs. In tables 1 and 2 and in the other tables to follow all doses are expressed in mg per kg of base.
As pointed out earlier, the introduction of the N-pbenethyl substituent enhanced analgesic potency, 2.6 to 8 times in mice, 6.3 times in rats and 2.5 times in rabbits. Contrary to the results in the morphine series, the substitution of phenacyl for methyl on nitrogen of (-) 3-hydroxymorphinan also enhanced analgesic potency, slightly more than phenethyl in mice, but similarly to phenethyl in rats and to a less degree than phenethyl in rabbits. By all tests activity was not increased as much by the introduction of N-(2-hydroxy-2-phenyl-ethyl) as by N-phenethyl, and analgesic activity was decreased materially below that of the N-methyl analogue when the substituent was phenoxyethyl. Also, analgesic potency was greatly reduced when the substituent on nitrogen was benzyl, phenylpropyl, 2-phenylbutyl, 4-phenylbutyl or 5-phenylamyl. The effect of the substituent N-phenylallyl presented one of the unusual variations from one set of observations to another. This compound, Ro 4-0556, had a low order of analgesic effect in mice, apparently no analgesic action in rats, but an MED 50 of 1.6 mg in rabbits, an effectiveness approximately one-third that of levorphanol. Ro 4-0554, in which the substituent was phenylpropargyl, had no analgesic action in mice or rats; it was not tried in rabbits. The effect of the addition of a substituent on the benzene ring of N-phenethyl in this series varied with the nature of the substituent and with the position of its substitution, though such variations were most apparent in the Bethesda observations. NH 2, NO 2, SCH 3 and CH 3 in the para-position, as well as 3,4-methylenedioxy, enhanced analgesic effectiveness in mice; para-N (CH 3) 2, OH or OCH 3 did not do so. Only para-NH 2increased analgesic effect in rats, while all substituents on the benzene ring diminished analgesic effectiveness in the rabbit. Addition of a substituent in the meta- or ortho-position or addition of more than one substituent on the benzene ring of N-phenethyl tended to reduce analgesic action. Curiously, SCH 3, although it increased analgesic potency in mice when added in the para-position of N-phenethyl, reduced such activity in all three species, mouse, rat and rabbit, when added in the para-position of N-phenacyl.
The effect of changing the aryl group in this series of N-aralkylmorphinans was very interesting. Most striking was the observation that complete saturation of the aryl groups, from phenyl to cyclohexyl or from pyridyl to piperidyl, very markedly reduced or practically abolished analgesic effectiveness. The compound containing N-morpholinoethyl was also very poorly effective. N-cyclohexenylethyl was a more effective substituent than N-cyclohexyl, but less effective than N-phenethyl. The compound having a 4-pyridylethyl group on nitrogen had a greater analgesic effect than the N-phenethyl derivative in mice, but not in rats or rabbits, whereas the presence of N-2-pyridylethyl gave a less potent analgesic agent than N-phenethyl in both mice and rabbits. The most potent compounds in the whole series in all three species were those in which the substituent on nitrogen was furylethyl (Ro 4-1539) or thienylethyl (Ro 4-1540). The former was 150 times more potent than levorphanol [(-) 3-hydroxy-N-methylmorphinan] in rats and 30 to 50 times more potent than levorphanol in mice and rabbits. The enhancement of analgesic activity by the substitution of thienylethyl was a little less than with furylethyl as the substituent.
In the four pairs of compounds which had the same substituent on nitrogen but in which one had hydroxy and the other methoxy in the 3-position, the methoxy was much weaker than the hydroxy derivative, as is the case in the methorphan-levorphanol pair and in the codeine-morphine pair. This was true for all observations on all three species. Removal of the 3-hydroxy group, as in Ro 1-5313 and Ro 1-5431, decreased analgesic effectiveness even more than muzzling of the hydroxy with a methyl group. This appeared to be the case also with Ro 4-1123, Ro 4-0774, Ro 4-1124 and Ro 1-9374, although in each of these instances the comparison was between a racemate and a levoisomer. The difference was so great, however, that there could be no doubt as to the direction of the change.
The one quaternary salt examined exhibited a much reduced analgesic action (Ro 4-0472, the methobromide, versus Ro 1-8439, the hydrobromide, of (-) 3-hydroxy-N-phenethylmorphinan), as had been observed previously in the morphine series (Eddy, 1933). The one methane sulfonate, perhaps owing to increased solubility, had twice the analgesic potency of the corresponding hydrochloride, (Ro 4-0288/1), the methane sulfonate, versus Ro 4-0288, the hydrochloride, of (-) (3-hydroxy-N-phenacylmorphinan). The methane sulfonate, however, was not more effective than the hydrochloride when the compounds were administered orally.
In the whole group of (-) N-aralkylmorphinans duration of effect was lengthened more often (20 times) than it was shortened (6 times) when comparison was made with levorphanol. It was, however, lengthened in three instances only when comparison was made with (-) 3-hydrox-y-N-ethyl-morphinan, which already had a significantly longer action than levorphanol.
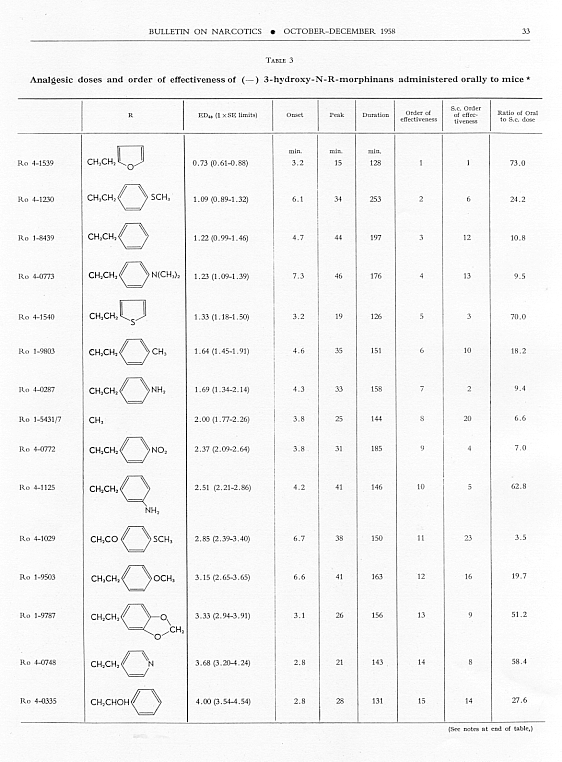
The order of analgesic effectiveness of the N-aralkylmorphinans was approximately the same for oral as for parenteral administration (see table 3), although there were some noteworthy exceptions. Both of the compounds having SCH 3 in the para-position of the benzene of the N-aralkyl group had a higher order of effectiveness in the series for oral than for parenteral administration. The N-phenethyl compound, Ro 1-8439, also stood higher, while the N-phenacyl compound, Ro 4-0288, was lower in order of effectiveness orally than parenterally. There was a reversal in the relative position of the pair, N-p-aminophenethyl- and N-p-dimethylaminophenethyl-morphinan. The former was relatively less and the latter was relatively more effective orally, whereas parenterally the N-p-aminophenethyl compound was relatively the more effective. Only seven compounds were more effective than levorphanol orally; nineteen were more effective parenterally. Only three compounds had a better ratio of oral to parenteral ED 50 than levorphanol. One of these, Ro 4-1029, was a good analgesic in mice, almost as good as morphine, but with one-seventh the activity of levorphanol. The N-ethyl analogue of levorphanol (Ro 4-1629), while a much weaker analgesic in animals, had an oral paren teral ratio of 2.0. The third compound (Ro 1-9543) with a good oral-parenteral ratio was a relatively weak analgesic by both routes.
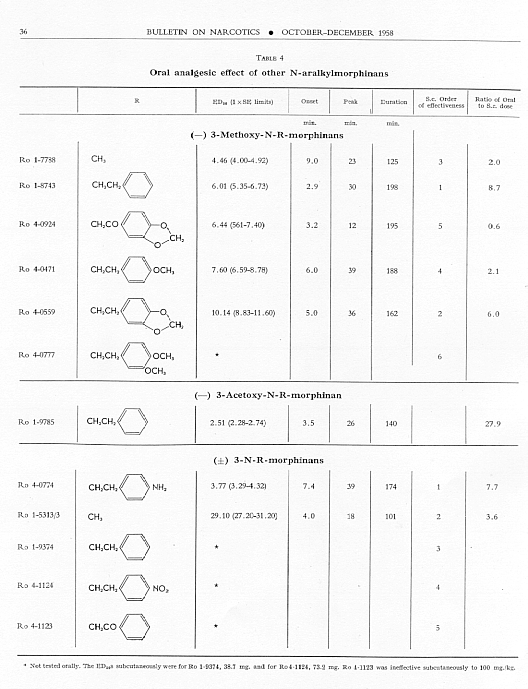
Of the five (-) 3-methoxy-N-aralkylmorphinans (see table 4) none was a better analgesic orally than (-) 3-methoxy-N-methylmorphinan, but all had relatively low oral-parenteral dose ratios. Ro 4-0924 [(-) 3-methoxy-N-(3,4-methyl-enedioxyphenethyl)-morphinan] had an oral ED 50 lower than the parenteral; it was, however, a weak analgesic by either route.
The duration of analgesic action was almost always longer after oral than after subcutaneous administration, without much or a consistent change in onset of effect.
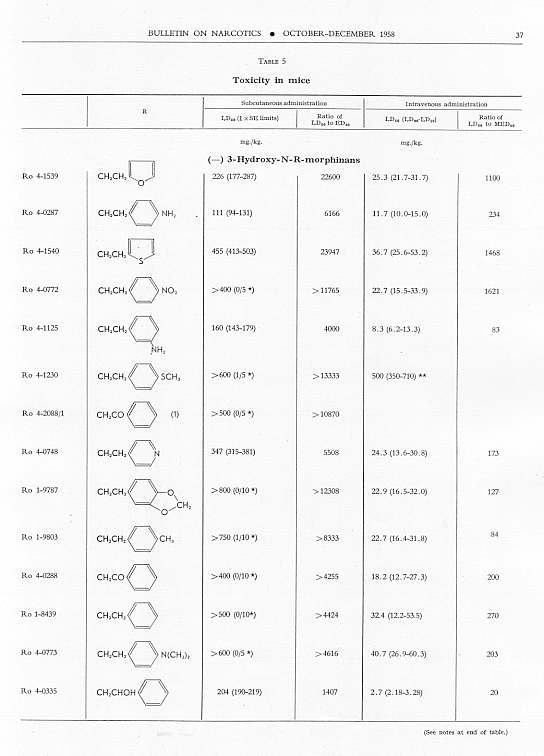
Toxicity.Data on toxicity, subcutaneously and intravenously in mice, are summarized in table 5, and ratios have been calculated for
|
S.c. LD 50 |
and |
I.v. LD 50 |
|
S.c. ED 50 |
|
S.c. MED 50 |
Allmost all the N-aralkyl derivatives had a lower subcutaneous toxicity than levorphanol, so that many of them had very high LD 50: ED 50 ratios. These ratios diminished as the analgesic activity diminished. The most toxic compounds were those with NH 2or NO 2 in para- or meta-position on the benzene of the N-aralkyl subsituent. However, for the most toxic, Ro 4-0827. the ratio of LD 50 : ED 50 was still very much greater than that for levorphanol or for morphine. The intravenous LD 50 values varied much less from the LD 50 of levorphanol than did thesubcutaneous LD 50’s, but again the most active compounds had high LD 50 : MED 50 ratios; and again these ratios diminished as analgesic activity diminished.
Fifty-six compounds have been examined in which an aralkyl substituent has replaced methyl in the N-methyl-morphinan series; forty-two of these are (-) 3-hydroxy-N-R-morphinans, R being the aralkyl group. In the main the order of analgesic effectiveness for the series was the same for mice (two methods of observation), rats and rabbits. The most active compounds had furylethyl, p-aminophenethyl or thienylethyl, as the aralkyl substituent. The first was 30 and the other two slightly more than 15 times more potent as analgesics than levorphanol in mice. The potency of each of these three compounds was very much greater relative to levorphanol in both rats and rabbits.
If the aryl group was phenyl, the optimal alkyl group was ethyl. Analgesic effectiveness was very much reduced by partial or complete saturation of the aryl group, as in the change from phenyl to cyclohexenyl and cyclohexyl or from pyridyl to piperidyl. If a substituent was added to the aryl group it seemed preferable to have it in the para-position.
Only in a few instances among the aralkyl derivatives was the ratio of oral to subcutaneous analgesic effectiveness as good as or better than that of levorphanol.
Aralkyl substitution generally decreased subcutaneous toxicity; it had much less effect on intravenous toxicity. The ratio of LD 50, whether subcutaneous or intravenous, to ED 50 or MED 50 decreased as analgesic effectiveness decreased.
BRAENDEN, O. J., EDDY, N. B. & HALBACH, H. (1955) Bull. Wld Hlth Org. 13, 937.
EDDY, N. B. (1933) J. Pharmacol. 49, 319.
EDDY, N. B. & LEIMBACH, D. G. (1953) J. Pharmacol. 107, 385.
GORDONOFF, T. & RUCKSTUHL, K. (1939) Dissertation,Bern.
GROSS, F. (1947) Helvet. Physiol. et Pharmacol. Acta, 5, 31.
KOLL,W. & REFFERT, H. (1938) Arch exper. Pathol. PharmakoI. I90, 687.
WINTER, C. A., ORAHOVATS, P. D. & LEHMAN, E. G. (1957) Arch. internat, pharmacodyn. 110, 186.
Eric Einar Ekstrand died on 24 August 1958 in Nykoeping, Sweden, his native town. He was one of the pioneers in the field of international narcotics control.
Born on 22 December 1880, he was a brilliant scholar and became a doctor of law of the University of Upsala. He then joined the Ministry of Foreign Affairs, where he occupied several positions, and became Director of Administrative Affairs in 1921. At the end of the first world war, he had begun a series of international missions which took him to all parts of the world, wherever there was need for men of goodwill.
In 1921-22, the Swedish Red Cross engaged him to study the famine situation in the Samara district of the Ukraine; next, he was chief of the International Commission for the Exchange of Greek and Turkish populations (1923). In 1924, the League of Nations entrusted him with the task of supervising the protection of the Albanian minority in Greece.
In 1925, he returned to the diplomatic service and was appointed Envoy Extraordinary and Minister Plenipotentiary in Buenos Aires, Santiago, Montevideo and Asuncion. In 1929 and 1930, he was Chairman of the League of Nations Commission of Enquiry into the Control of Opium Smoking in the Far East. He travelled through south-east Asia studying closely not only the opium question, but all humanitarian problems.
Back in Europe in 1931, he became director of the section of the Traffic in Opium and Other Dangerous Drugs, and of the Social Section of the League of Nations and, ipso facto, secretary of the Advisory Committee on Traffic in Opium and Other Dangerous Drugs. The same year, he was secretary-general of the Conference for the Limitation of the Manufacture of Narcotic Drugs. This conference drafted the Convention for limiting the Manufacture and regulating the Distribution of Narcotic Drugs, one of the basic treaties for the international control of these substances. He was also secretary in 1936 of the Conference for the Suppression of the Illicit Traffic in Dangerous Drugs.
During the second world war, Eric Einar Ekstrand published his memoirs, Around the World in Thirty Years - a title which he thought at the time signified the conclusion of his career. In fact, he was to continue for years to come his untiring activity.
After having been a member of the unofficial Swedish Commission for assistance to Finland during the war of 1939/40, he returned to the international field as chairman for six years of the United Nations Sub-Committee for the Prevention of Discrimination and the Protection of Minorities.
When he died, after a long illness, it marked the end of a life of service and devotion to duty.
The Editors of the Bulletin on Narcotics extend their sincere sympathy to Mr. Ekstrand's family.