I. SYNTHETIC STUDIES DIRECTED TOWARDS THE PREPARATION OF ANALGESIC COMPOUNDS
II. SYNTHETIC INTERCONVERSIONS OF MORPHINE DERIVATIVES
III. THEBAINE ADDUCT WITH 1,4-NAPHTAQUINONE
Author: David Ginsburg
Pages: 1 to 4
Creation Date: 1959/01/01
The last review article in this series covered work published in the chemical literature through most of 1957 ( [ 1] ). The papers reviewed in the present article may be divided into several types: ( a) Synthetic analgesics structurally related to the morphine alkaloids; ( b) Interrelationships among various morphine derivatives; ( c) A continuation of the structural elucidation of Diels-Alder adducts of thebaine.
An extremely interesting account has been published by Gates & Webb ( [ 2] ) describing the synthesis of 3-hydroxy-N-methyl- isomorphinan (I) and of 3-hydroxy-Δ 6-dehydro-N-methyl- isomorphinan (II).
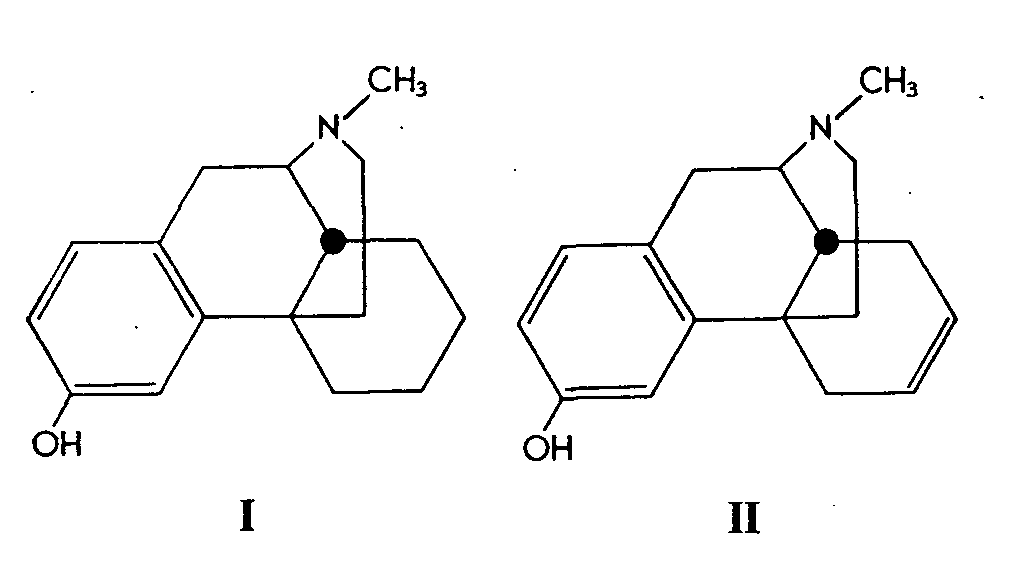
Both of these synthetic substances were resolved, and the l-isomer of each was shown to possess powerful analgesic activity. l-3-Hydroxy-N-methyl- isomorphinan (I) is 8 to 10 times as active as morphine as determined by the D'AmourSmith tail-flick method, whilst the l-isomer of (II) is only slightly less active.
The synthesis of these compounds extends the method developed by Gates in his total synthesis of morphine, thus increasing the number of valuable products which may be obtained by the first successful synthetic approach to this complex natural product. Here also a naphthaquinone derivative reacted with butadiene and afforded rings A, B, and C of the morphine skeleton. Subsequent cyclization of the lactam ring afforded the tetracyclic skeleton of the alkaloid.
The resolution of the synthetic compounds (I) and (II) was effected with the enantiomorphic dibenzoyltartaric acids. Because of synthetic difficulties, (II) was obtained via the O-methyl ether, which was later cleaved by potassium hydroxide to afford the corresponding phenol.
3-Hydroxy-N-methyl- isomorphinan (I) could also be obtained by nitration of the lactam, 16-oxo-N-methyl- isomorphinan (IV) which, in turn, was obtained by methylation of 16-oxo- isomorphinan (III) with methyl iodide in the presence of sodium hydride.
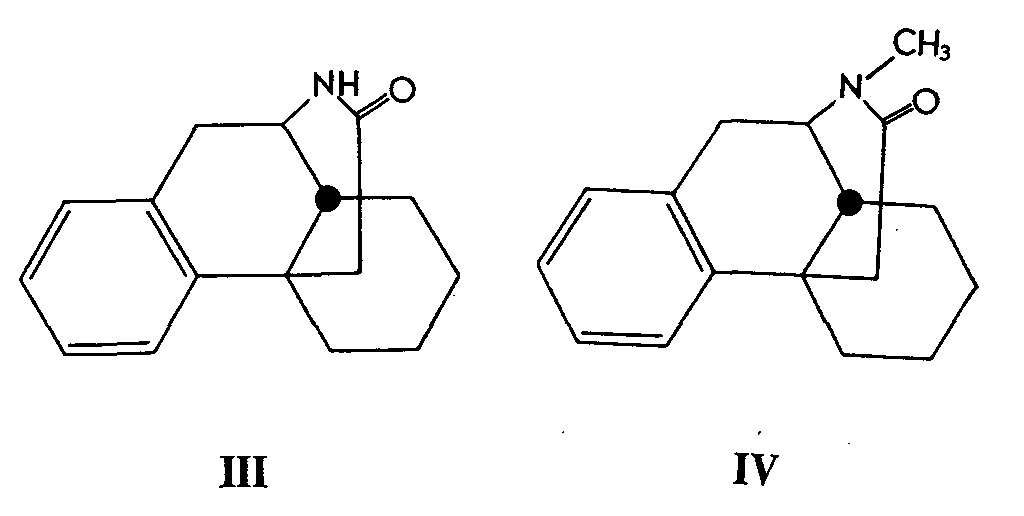
Nitration of (IV) gave a mixture, containing preponderantly the 3-nitro-isomer (V), which was accompanied by lesser amounts of the 1- and 2-nitro derivatives. All three isomers were separated and each was converted in a series of steps into the corresponding (1-, 2-, and 3-) amino derivatives. Diazotization and hydrolysis converted each amine of type (VI) to the corresponding phenol (I and 1- and 2-hydroxylic isomer).
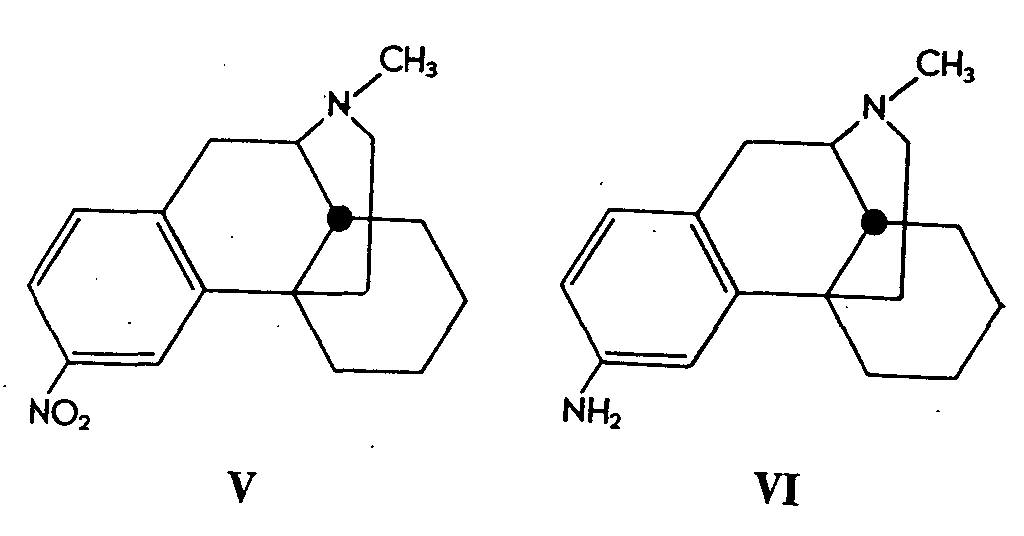
The structural assignments for the minor isomers were made on the basis of infrared analogies to suitably substituted benzene derivatives and attack (by nitration) of the 4-position was reasonably ruled out on the basis of both steric and electronic considerations.
Although a new compound (VII) prepared by May ( [ 3] ) does not possess the skeletal structure of morphine, it is felt that it falls within the scope of this review for two reasons. It was prepared in order to determine whether it would possess analgesic activity and it is a positional isomer of 3-hydroxy-N-methyl-morphinan (VIII).
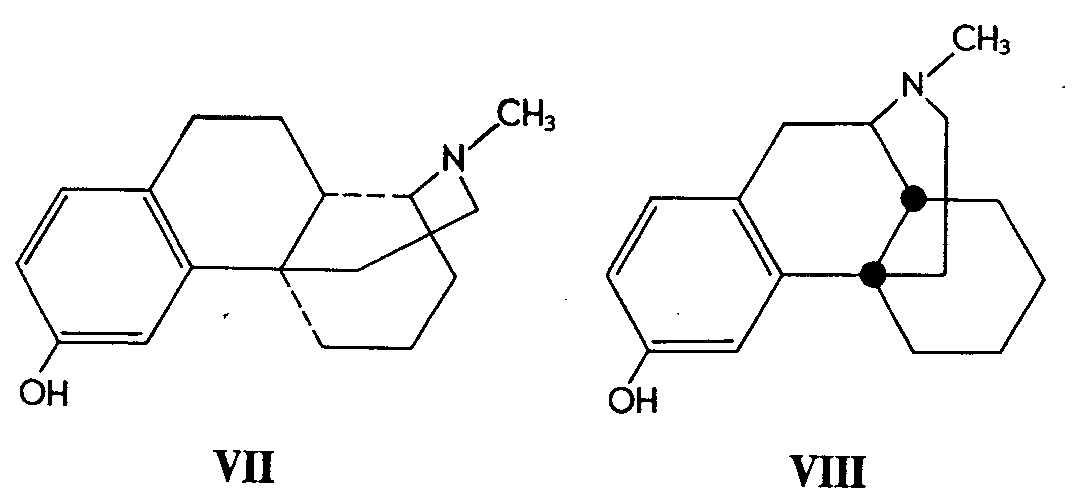
The new compound, 1,2,3,9,10,10a-hexahydro-6-hydroxy-11-methyl-1,4a (4H)-iminoethanophenanthrene (VII) had less than 2% of the analgesic potency of racemic 3-hydroxy-N-methyl-morphinan (VIII).
Morphinone (XI) is of interest, since it may be postulated that it is a biogenetic precursor of certain morphine alkaloids - e.g., morphine itself could be formed by reduction of (XI), codeine could be formed by reduction and methylation and thebaine would result by dehydrogenation and dimethylation (4).
Rapoport and his co-workers (5) have recently prepared this sensitive ketone (XI) by oxidation of morphine whose phenolic group was protected with a methoxymethyl group (IX), by means of silver carbonate in benzene. The protecting group still present in (X) could be removed under mild acidic conditions or on a sulfonic acid type ion exchange resin. Elution from the resin must be carried out under very-well-defined conditions in order to avoid the destruction of (XI) by alkali towards which it is extremely sensitive.
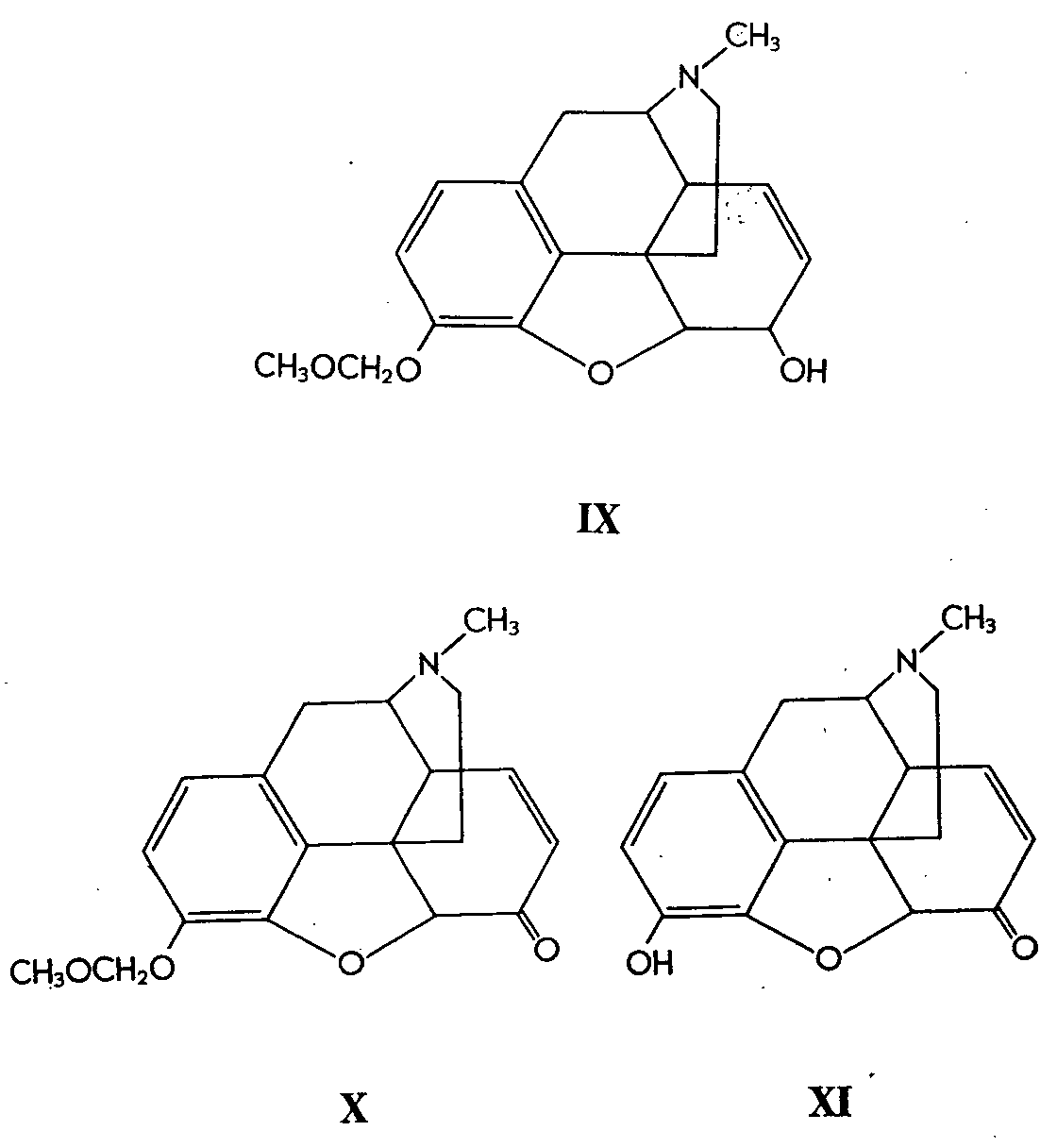
Reduction of morphinone with sodium borohydride was possible and gave morphine. Its reduction with 5% palladized carbon in ethanol gave dihydro-morphinone (XII).
Treatment of (X) with methyl lithium followed by hydrolysis of the methoxymethyl protecting group gave 6-methylmorphine which upon methylation could be converted into 6-methylcodeine.
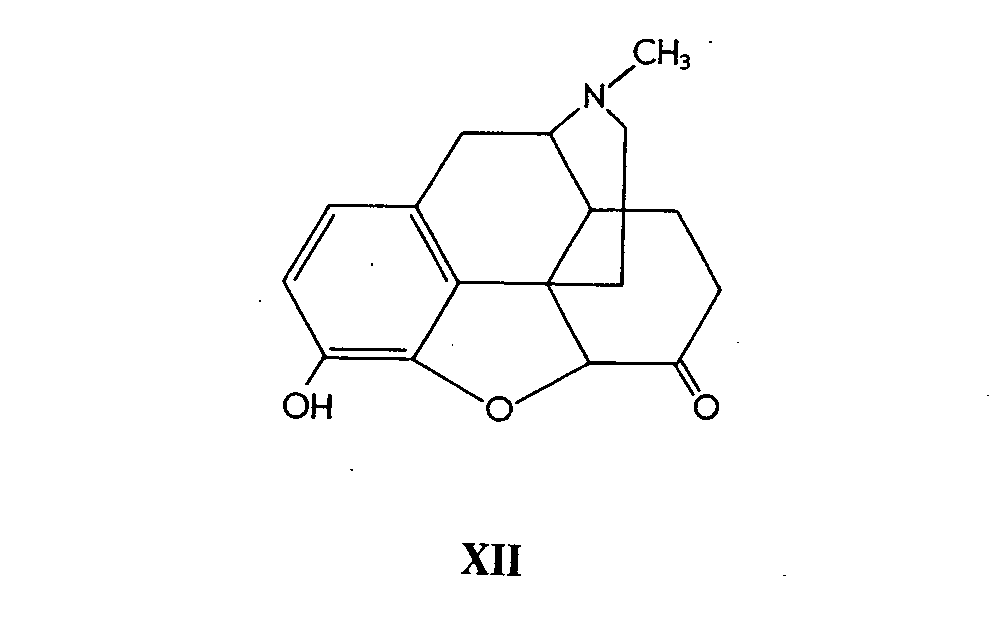
Weiss ( [ 6] ) has shown that 14-hydroxycodeine (XIII) can be demethylated with aqueous hydrobromic acid to afford 14-hydroxymorphinone (XIV). (No skeletal rearrangement occurs, since the 14-position is blocked.) (XV) was shown to accompany (XIV) as a reaction product, but the co-appearance of 8,14-dihydroxydihydromorphinone (XV) was shown to be due to a secondary reaction. It is formed from (XIV) by addition of hydroxyl ion during the alkaline isolation step. It would be of interest to correlate the stucture of (XV) to that of (XVIII) (see below) by direct interconversion reactions.
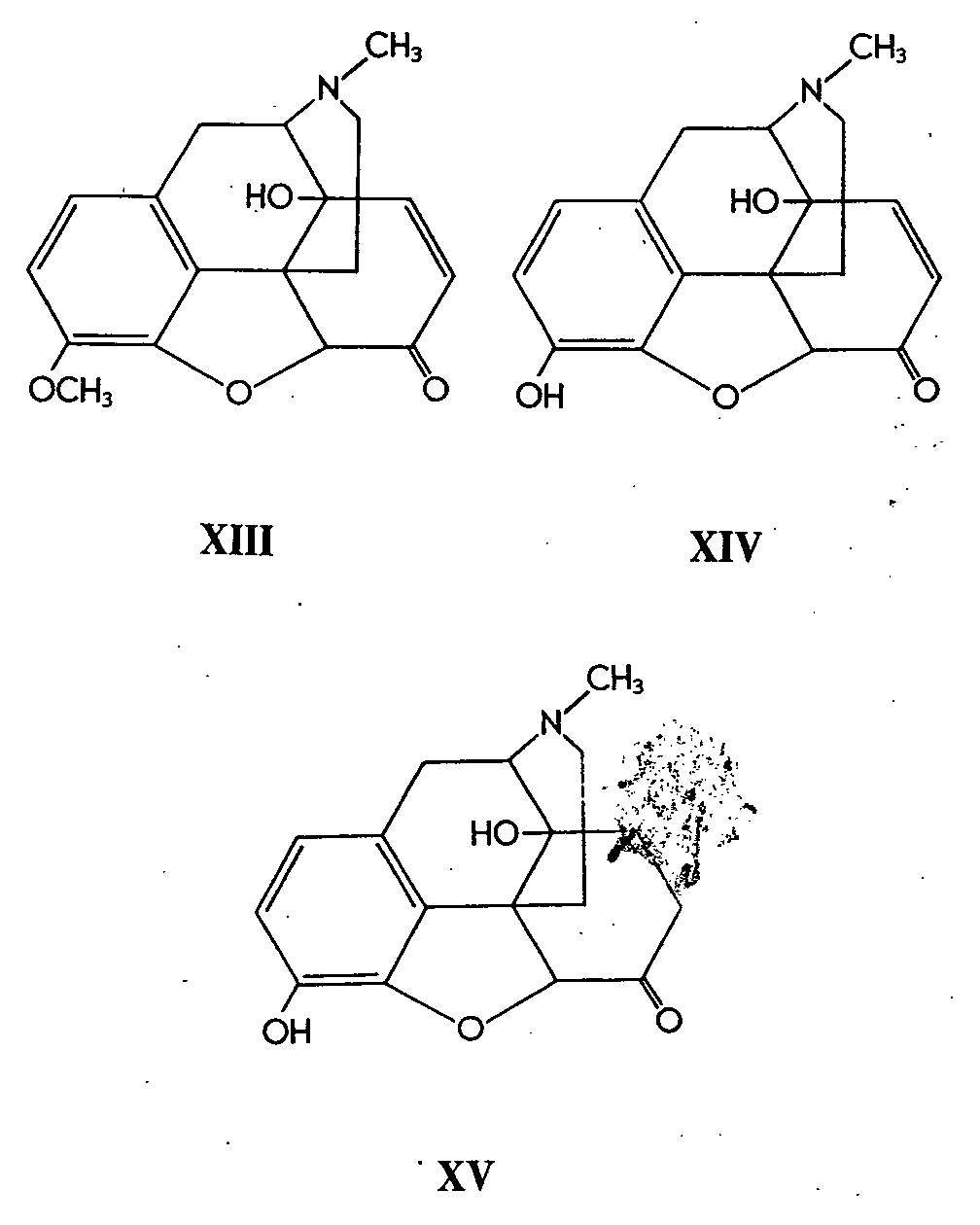
Sargent, Schwartzman & Small ( [ 7] ) have reported the preparation of a series of hydroxylated codeine and neopine derivatives by treatment with osmium tetroxide and-decomposition of the osmate osmate esters. Although the hope that these products might be pharmacologically active was not realized, several steric conclusions were drawn from this work.
For example, osmium tetroxide hydroxylation of acetylneopine (XVI) gave a presumed cis-glycol (XVII). Viebock (8) had previously obtained 8, 14-dihydroxydihydrocodeinone (XVIII) by oxidation of thebaine with manganic acetate followed by treatment with hot dilute hydrochloric acid.
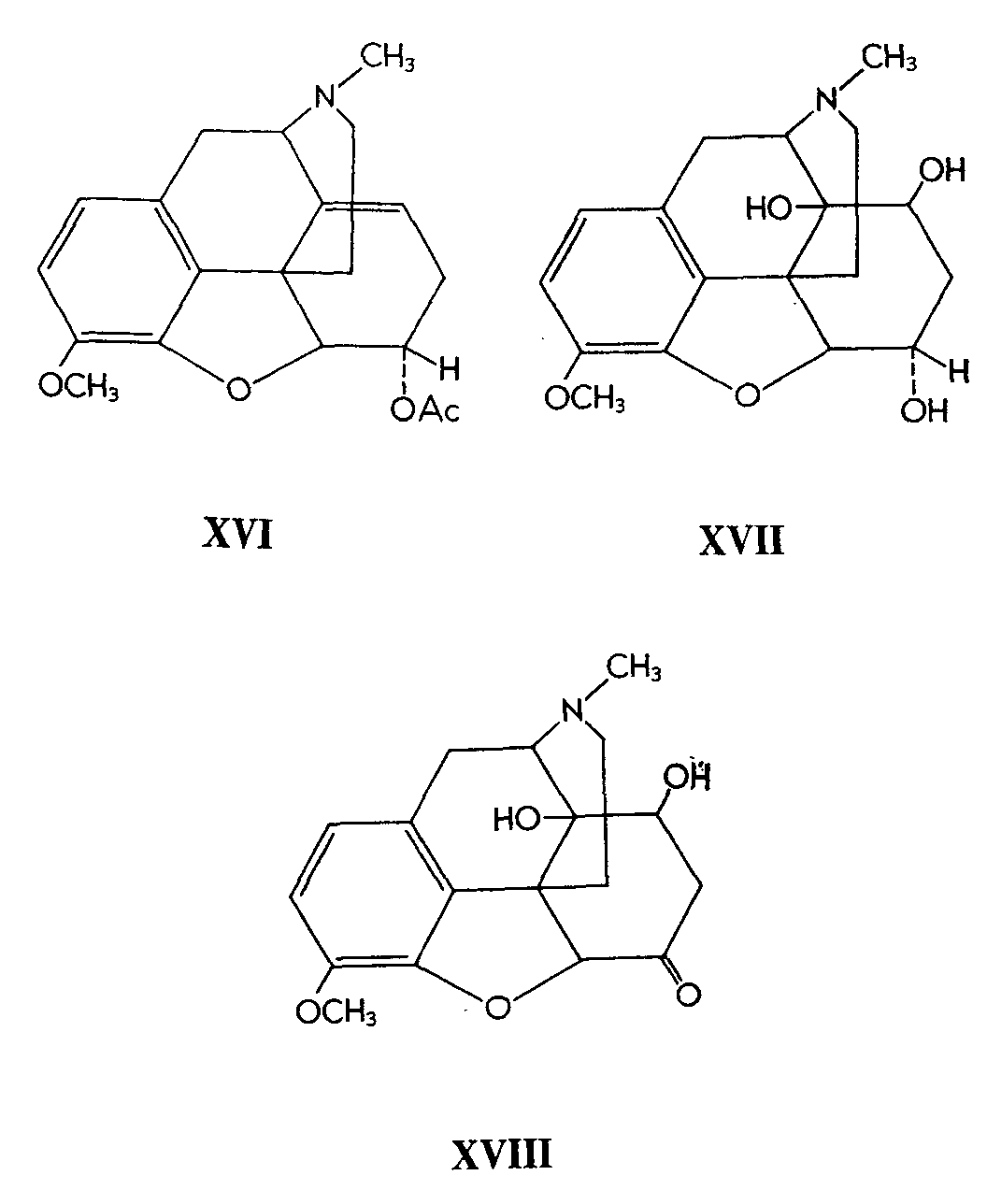
Reduction of Viebock's ketone (XVIII) with lithium aluminium hydride or with sodium borohydride gave a mixture of epimeric (at C 6) 8,14-dihydroxydihydrocodeines. One of these was identical with (XVII), proving that the 8-and 14-hydroxyl groups are disposed in the cis-fashion. In the other epimer, the authors assume that the apposite configuration of the hydroxyl obtains at C 6.
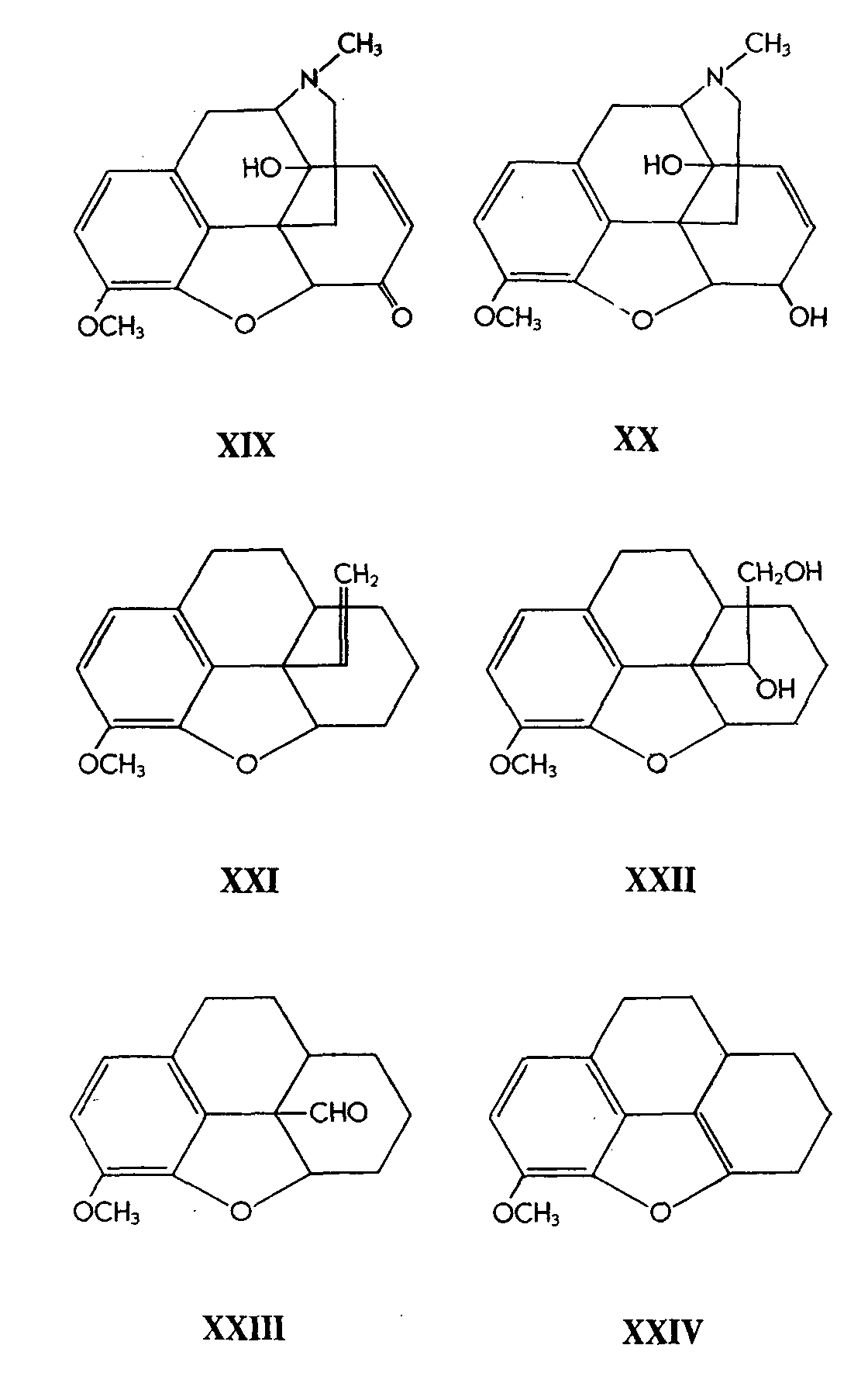
14-Hydroxycodeine (XX) was prepared by sodium borohydride reduction of 14-hydroxycodeinone (XIX). By analogy to the stereospecific reduction of codeinone to codeine by means of this reducing agent (9), the authors infer that (XX) has the codeine configuration of the hydroxyl group at C 6 and that its reduction product, 14-hydroxydihydrocodeine-B has the similar configuration at C 6. The C 6-epimer of the latter, 14-hydroxydihydrocodeine-C is therefore assumed to have the isocodeine configuration. It is a pity that the authors have not proved this point conclusively by studying the rates of hydrolysis- e.g., of the acetates of the pair of epimers 14-hydroxydihydrocodeine-B and-C (cf. 10).
Rapoport and co-workers ( [ 11] ) have shown that aldehyde degradation products of codeine very readily lose carbon monoxide to give a benzofuran derivative- e.g., 6,7,8,9,10, 14-hexahydromorphenol methyl ether.
Codeine was converted via its tosylate to Δ 7-desoxycodeine through treatment of the tosylate with lithium aluminum hydride. Degradation to the methine and to the 13-vinyl derivative (XXI) was effected in 52% over-all yield from codeine, since there was no hydroxyl group at C 6 to lower the yield through interaction during the Hofmann degradation steps. The vinyl compound was quantitatively hydroxylated by means of osmium tetroxide, and the resulting glycol (XXII) was cleaved to the aldehyde (XXIII) by means of sodium periodate. The aldehyde was very readily decarbonylated through a free radical mechanism under a variety of reaction conditions to yield the substituted benzofuran (XXIV).
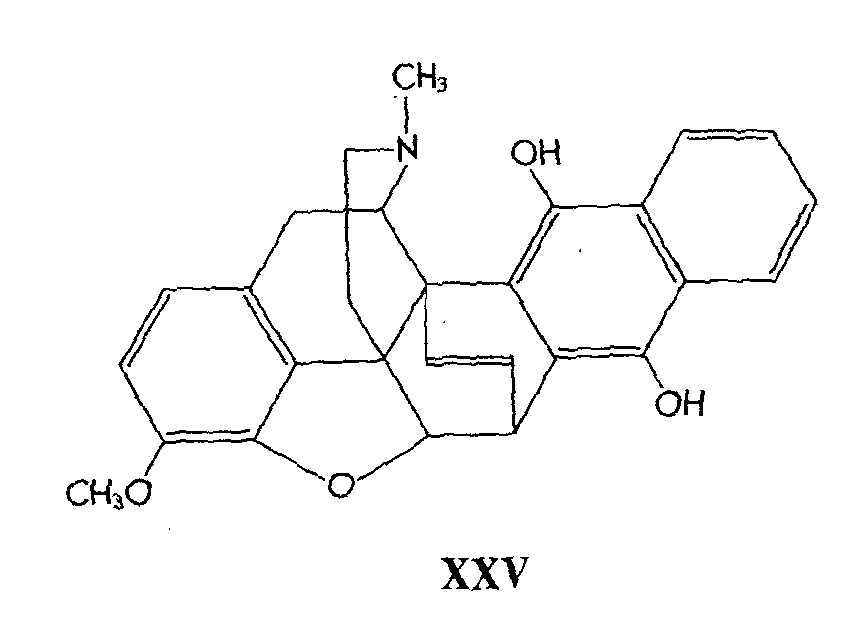
Bentley, Ball & Cardwell (12) have published an additional paper in the field of Diels-Alder adducts of thebaine. The 1,4-naphthaquinone adduct of thebaine has been shown to parallel in its chemical behaviour that of the analogous benzoquinone adduct.
The naphthaquinone adduct is unstable, and cannot be isolated as such. It was readily isomerized to thebaine1,4-naphthaquinol (XXV).
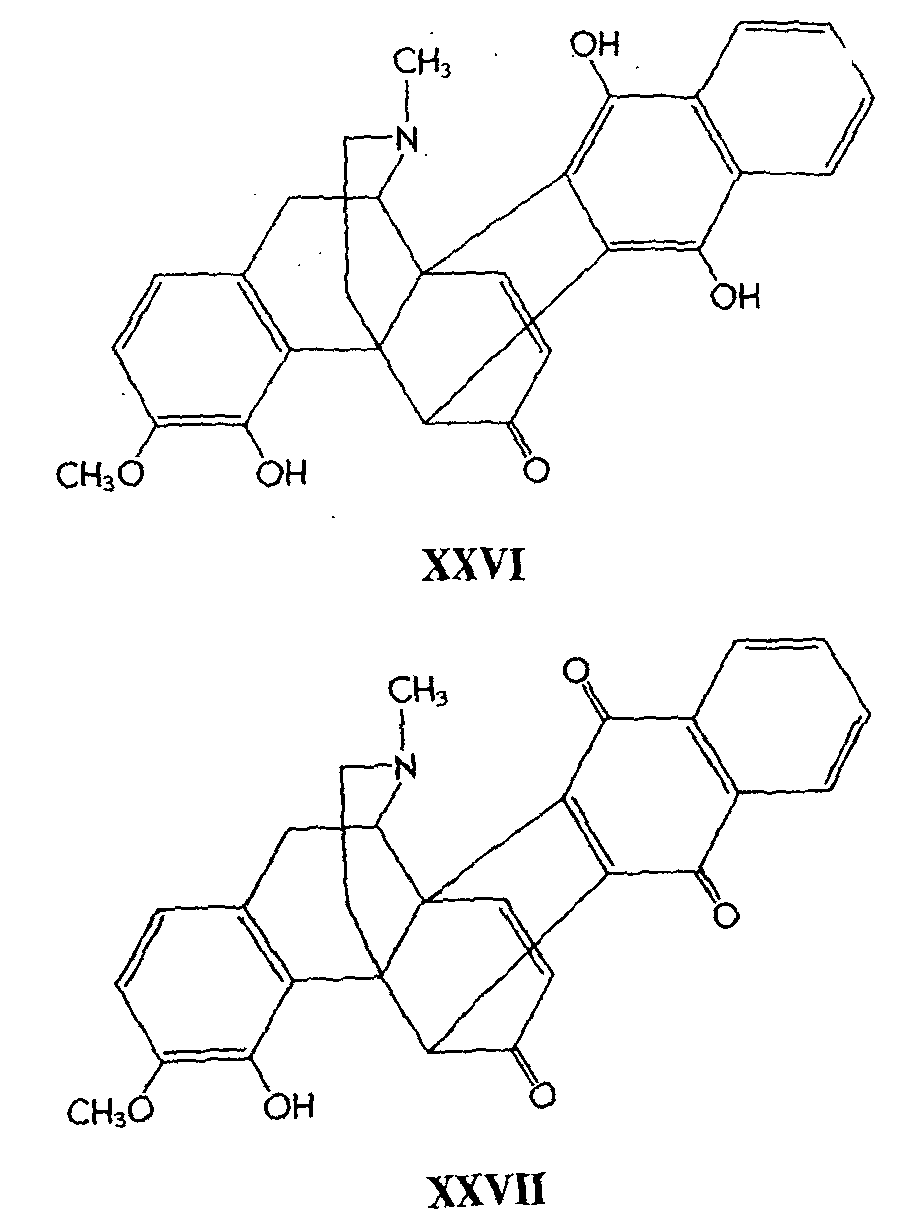
The acid rearrangement product, benzflavothebaone (XXVI), is analogous to flavothebaone, but in contradistinction to the latter it is very readily oxidized by air in alkaline solution to the quinone (XXVII).
GINSBURG, Bulletin on Narcotics, Vol. X, No. 2, 1 (1958).
002GATES & WEBB, J. Amer. Chem. Soc., 80, 1186 (1958).
003MAY, J. Org. Chem., 23 , 947 (1958).
004Cf. SCHOPF, Naturwiss., 39, 241 (1952).
005RAPOPORT, BAKER & REIST, J. Org. Chem., 22, 1489 (1957).
006WEISS, J. Org. Chem., 22, 1505 (1957).
007SARGENT, SCHWARTZMAN & SMALL, J. Org. Chem., 23, 1247 (1958).
008VIEBOCK, Ber., 67, 197 (1934).
009GATES, J. Amer. Chem. Soc., 75, 4340 (1953).
010ELAD & GINSBURG, J. Amer. Chem. Soc., 78, 3691 (1956).
011RAPOPORT, BATCHO & GORDON, J. Amer. Chem. Soc., 80, 5767 (1958).
012BENTLEY, BALL & CARDWELL, J. Org. Chem., 23, 941 (1958)


