A. Main types of products on the Swedish market
B. Organization of the National Drug Control Authorities
C. Basic principles of drug control
D. Registration of pharmaceutical specialities
E. Ad interim sales permits
F. Market control and re-examination of registration
G. Prescription and classification according to the royal statute on poisons
H. Narcotics control measures
Author: Leonard Goldberg, Gunnar Lindgren
Pages: 7 to 15
Creation Date: 1961/01/01
For many years drugs to be introduced on the market in Sweden have been submitted to strict control measures. These apply to chemical compounds used as drugs as well as to biological products, to single substances as well as to mixtures or compositions, including narcotic and habit-forming drugs. The effect of the existing laws has been to facilitate the introduction of new products of value, to protect the public from using drugs of doubtful or no value, and to keep down the cost of medication.
The aim of this study is to give a survey of the existing lesgislation, its administration and general effects.
The major portion of the retail sale of medicines in Sweden is monopolized and is limited to the pharmacies, about 540 in all, corresponding to one pharmacy for 14,000 individuals.
For distribution of certain necessary medicines in less populated areas, about 800 distribution centres have been set up, connected to the pharmacies and keeping in stock a limited number of medicines not on prescription. The pharmacies, which are owned privately, co-operate very closely and have formed a financial and administrative union (Apotekarsocieteten) with strict and detailed rules for all the members of the organization, approved by the Ministry of the Interior.
The turnover of the pharmacies in 1960 was about 400 million crowns, two-thirds of this sum being for sales on prescription. Eighy-five per cent of the prescriptions comprised pharmaceutical specialities (proprietary products) and "standardized" galenic preparations (see below), the rest being ex tempore preparations.
Cosmetics, some common preparations for fresh wounds, hair lotions, certain saline laxatives, liniments and some vitamin preparations may also be sold outside the pharmacies. These products are controlled by a special council, supervising the advertising of these products. The National Board of Health appoints one of the members of this council. The other members are from the press, industry and the advertising field. The council operates mainly in an advisory capacity, but has the means to stop misleading advertising in the newspapers, magazines and other periodicals.
1Professor of research on alcohol and analgesics, Karolinska Institutet' Stockholm; expert member (pharmacology) of the Council on Drug Acceptance of The Swedish National Board of Health; expert member of the Advisory Panel on Addiction-Producing Drugs of the World Health Organization.
2Professor of Social Medicine, University of Lund; expert member (internal medicine) of the Council on Drug Acceptance of the Swedish National Board of Health; former head of the drug and remedy section of the Swedish National Board of Health.
The products sold by the pharmacies comprise pharmaceutical specialities (proprietary products) and other preparations, mostly galenicals.
Pharmaceutical Specialities (Proprietary Products)
The greater part of the medicines sold in Swedish pharmacies are pharmaceutical specialities (proprietary brands). By these are meant preparations sold to the user in the container in which they are supplied by the manufacturer. The sale of these products is controlled by the royal ordinance concerning trade in pharmaceutical specialities of 15 June 1934 (Specialitetskungörelsen).
Prior to the introduction of this ordinance, the market was flooded with so-called patent medicines, many of extremely doubtful value. The introduction of the regulations in 1934 resulted in a much needed clean-up in this field.
The number of specialities on the Swedish market in recent years has been between 2,000 and 3,000, which is considerably less than in most other countries.
About half of the pharmaceutical specialities are of Swedish origin: the rest are imported. The number of remedies accepted for sale has varied somewhat from year to year, and shows a slight tendency to increase (fig. 1).
All addiction-producing drugs, covered by international treaties, are also controlled in Sweden. The number of addiction-producing drugs actually sold on the Swedish market and covered by international treaties is, however, considerably less (14 only), distributed among 62 proprietary products, containing one drug only or compositions.
The narcotics sold on the Swedish market include, besides opium, the following opium derivatives: morphine, codeine, ethylmorphine, hydrocodone, hydromorphone and oxicodone. Among synthetic drugs with morphine-like properties, 8 drugs are sold: dextromethadone, dextromoramide, diethylthiambu-tene, ketobemidone, methadone, normethadone and pethidine.
Some preparations containing codeine in combination with other active ingredients are exempt from prescription, while preparations containing ethylmorphine plus an active ingredient are exempt from narcotics control, but are on prescription.
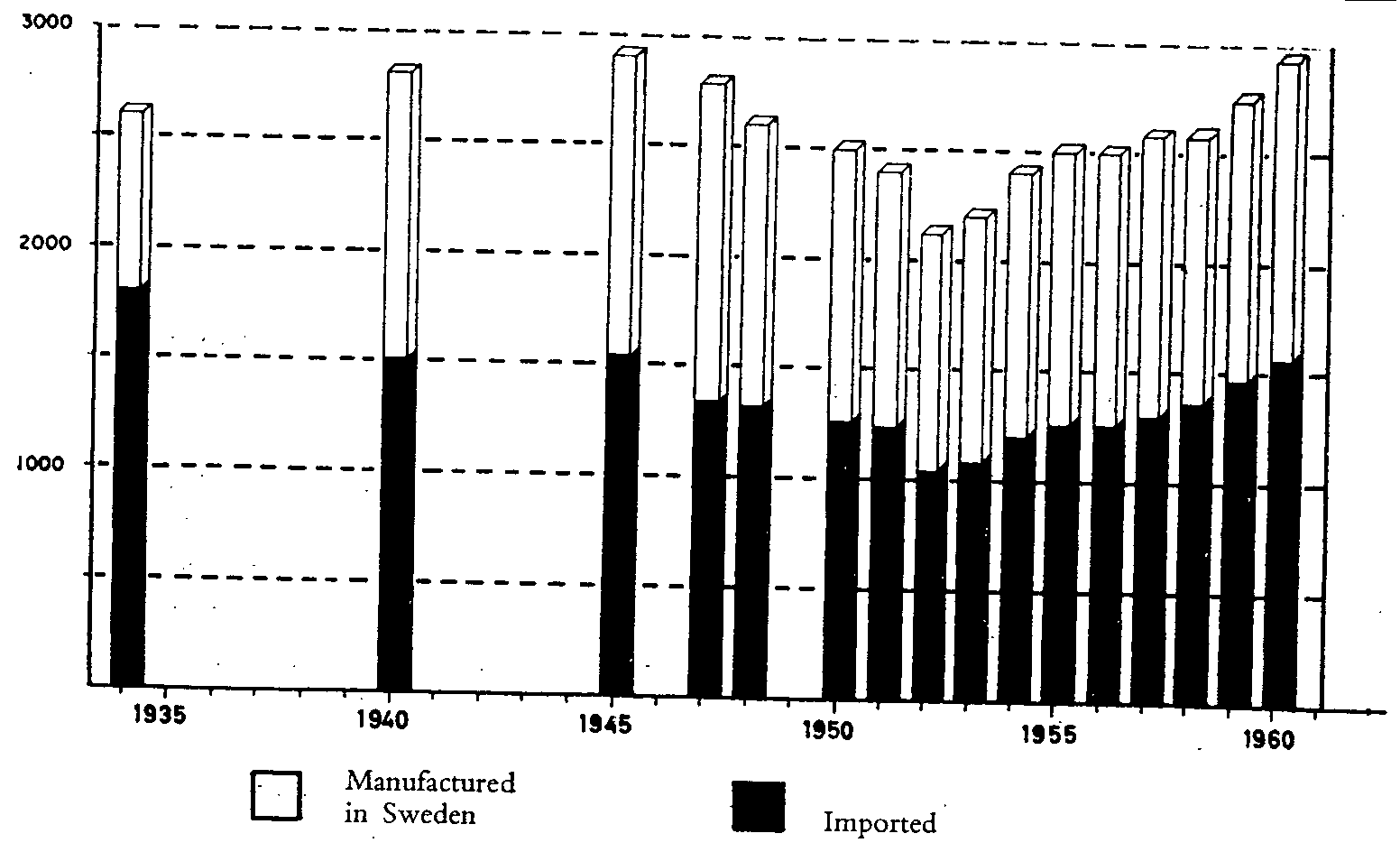
Number of pharmaceutical specialities, registered or sold on general ad in terim sales permit in Sweden, 1934-I960
Exempt from narcotics control are also dextromethorphan and pholcodine.
Cocaine is used as a local anaesthetic only in exceptional cases, and delivered as an ex tempore preparation.
Stimulating drugs on the Swedish market which are likely to be abused - e.g., agents used therapeutically for obesity, but abused to a growing extent especially by juveniles - and not covered by international treaties, can be designated as narcotics under existing Swedish laws. Seven stimulating drugs distributed among 20 proprietary products on the market have been declared narcotic drugs - viz., acetyloxiphenylmethyl piperidine, amphetamine, dexamphetamine, metamphetamine, methylphenidate, phenmetraline and pipradol.
The number of analgesic drugs not on prescription - mainly drugs with antipyretic properties - is about 60.
The number of substances on prescription with sedative, tranquillizing or ataractic properties is about 50, distributed among about 210 proprietary brands.
Other Preparations (including Galenics)
Each pharmacy is equipped to produce a composition ex tempore according to an individual prescription. As a rule an ex tempore galenic preparation is prepared in the pharmacy to which the prescription is delivered. Within the pharmaceutical organization, however, there is considerable manufacture of those non-proprietary products which do not deteriorate easily, can be kept in stock indefinitely, and have a "standardized "composition. This manufacture is confined to 18 district laboratories connected with certain pharmacies and equipped for manufacture on an industrial scale.
1.The National Board of Health
The National Board of Health (Kungl. Medicinalstyrelsen), headed by a director-general, is the executive and administrative organ of the Swedish Ministry of the Interior for questions regarding public health.
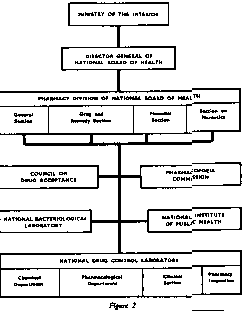
The board comprises a number of administrative units divided into sections. The unit managing drug control and the supervision of the pharmacies is the pharmacy division, divided into four sections (fig. 2).
The general section supervises the quality and the manufacture of medicines in the pharmacies and in the district laboratories.
The drug and remedy section has the responsibility for the introduction and control of proprietary products, and handles questions regarding the import, export and sale of pharmaceutical specialities.
The financial section supervises and confirms the prices of the raw products and medicines that are manufactured and sold by the pharmacies. It also investigates the prices of the industrially produced preparations.
The narcotic section handles questions regarding narcotic drugs - i.e., import, export, sale and legal matters concerning these drugs.
A close co-operation exists between the different sections. The pharmacy division works more as a team of different specialists than as a unit split up into separate sections, the head of the general section being a pharmaceutical chemist, and the head of the drug and remedy section a physician and an expert in pharmacology. Legal experts are attached to the different sections.
The National Drug Control Laboratory (Statens farma-ceutiska laboratorium) has as its main task the proper control of drugs to be introduced on the market or already on the market.
The laboratory is directly connected with the National Board of Health, and is divided into three departments and one section (fig. 2).
The chemical department carries out chemical and pharmaceutical control as well as research within these fields, and advises in matters concerning the classification of substances according to the statute on poisons, and other national and international requirements.
The pharmacological department covers biological assay of pharmaceutical products and carries out research on the action and uses of drugs, including specific work on biological standardization - e.g., the preparation of biological standards.
A special section attached to the pharmacological department deals with problems of clinical pharmacology and experimental therapeutics, with special regard to assessing the clinical value of drugs intended for sale on the market and evaluating indications, possible side actions and claims regarding use of products.
The pharmacy inspection supervises the production of galenic preparations in the pharmacies and collects samples of drugs in the market which are to be analysed in the laboratory for purposes of market control.
Close co-operation exists between the National Drug Control Laboratory and other governmental or semi-governmental agencies. Assay of antibiotics and control of sterility of solutions intended for injection are handled by the National Bacteriological Laboratory. Vitamin analyses of pharmaceutical specialities are carried out by the vitamin department of the National Institute of Public Health. The control laboratory of the Swedish Pharmaceutical Society (AKL) is responsible for most of the control of raw materials and other products distributed to and used in individual pharmacies. This laboratory also edits the list of standard preparations prepared in the pharmacies and proprietary products allowed to be sold in Sweden.
The Council on Drug Acceptance (Specialitetsnämnden) advises the National Board of Health on the acceptance or refusal for sale of proprietary products.
The council consists of five voting members, all appointed by the Government. The chairman is an expert in jurisprudence, holding a prominent governmental position; the others are experts representing internal medicine, pharmacology, pharmacy and the chemical industry. The head of the drug and remedy section, as well as the representatives of the various departments of the National Drug Control Laboratory, participates in the discussions of the council, but has no right to vote.
In the case of new products to be introduced on the market, the council advises the National Board of Health whether the product shall be registered as a pharmaceutical speciality with the board and given a permanent sales permit, or whether other measures shall be taken. Other tasks of the council include consideration whether a product should be considered a pharmaceutical speciality or not, establishment of general principles regarding indications for use of special groups of substances, and special measures regarding advertising of narcotic products, etc. With regard to products already on the market and subject to control, the council can advise whether or not the product is to remain on the market.
The council may also be consulted in special cases concerning the classification of new products according to the statute on poisons or to national or international narcotics legislation.
The basic laws now in effect concerning the sale of drugs in Sweden are the royal statute on pharmaceutical products of 1913 [ 1] , the royal statute of poisons of 1943 [ 2] , the royal ordinance concerning the control of and trade in certain bacteriological preparations intended for human use of 1925 [ 4] , and the royal ordinance concerning trade in pharmaceutical specialities of 1934 [ 5] .
Regulations concerning addiction-producing drugs are to be found in the royal ordinance concerning narcotics of 1933 [ 3] . Besides these regulations, Sweden is bound by existing international treaties concerning addiction-producing drugs.
All ex tempore preparations according to a doctor's, a dentist's or a veterinary's prescription must be made by a licensed pharmacist. The ex tempore preparations are not controlled per se. Instead, the pharmacy inspectors have to check whether raw materials or other substances used for making the ex tempore preparations and stored in the pharmacies are in good condition and fulfil the requirements of the Swedish Pharmacopoeia. They also ensure that the arrangements for the preparation of the final products are satisfactory from the point of view of safety and the elimination of errors.
The production of galenic preparations by the district laboratories is controlled by the control laboratory of the Swedish Pharmaceutical Society (AKL). As a routine matter, samples are also taken in the market by the pharmacy inspectors and analysed at the National Drug Control Laboratory.
Due to the impossibility of controlling the industrial production of pharmaceutical products (proprietary brands) in the same manner as the pharmacy preparations - i.e., at the place of production - the control of the pharmaceutical specialities is based on the examination of the final product, before it is introduced on the market, and at regular intervals after its introduction [ 7] .
The introduction of new drugs is subject to sales permits granted by the National Board of Health.
The sales permits are divided into three categories:
The individual sales permit (licence), granted to a doctor, a veterinary surgeon or a dentist after special application;
The general ad interim sales permit; and
The permanent sales permit (registration).
The sales permits may if necessary be revoked by the National Board of Health.
As the sale of the pharmaceutical specialities is restricted to the pharmacies, which are under strict control by the government, the distribution of the proprietary products, their introduction and release as well as their prohibition for sale is effectively administered by the national drug control authorities.
The principal prerequisite for the sale of a pharmaceutical speciality on the Swedish market is the inclusion of the product in the register of the National Board of Health. Hence, the manufacturer or his representative must apply to the National Board of Health for registration of the preparation in order to be allowed to sell the product, submitting relevant material, including samples of the product to be sold.
After the application has been filed at the National Board of Health, the first task is to decide if the product or the preparation is "intended to prevent, ameliorate or cure disease or symptoms of disease in man or animals ", and falls under the ordinance concerning trade in pharmaceutical specialities.
The royal ordinance concerning trade in pharmaceutical specialities (1934) [ 5] states that the National Board of Health, when a compound or a preparation is intended for sale, must pay special attention to five major points:
Whether the statement of the character and composition of the speciality is correct;
Whether the speciality is appropriate from a pharmaceutical point of view;
Whether the speciality can prevent, ameliorate or cure disease or symptoms of disease in man or animals - i.e., is medically useful;
Whether the price of the speciality is fair;
Whether the claims made in the advertising material are justified.
After a first survey of all relevant data has been made by the Drug and Remedy Section of the Board, the material, including samples of the drug, is sent to the National Drug Control Laboratory, where the actual control is carried out.
The control measures at the National Drug Control Laboratory are intended to check the points as outlined above. In order to concentrate the studies on essential features, a procedure for each single drug to be controlled is drawn up at regular meetings between the head of the drug and remedy section of the National Board of Health and the staff of the National Drug Control Laboratory. The actual work carried out is' intended to obtain information on five major points:
Composition
A fundamental prerequisite for registration is a correct statement of the composition of the speciality. This applies to the principal active constituents as well as to any other substance present in the product. The individual constituents are tested qualitatively and quantitatively by chemical methods.
In those cases where chemical methods are not specific or sensitive enough, biological assay is carried out. The testing is made in comparison with reference standards, sent in by manufacturers in case of new substances or with the appropriate international biological standards when the drug is biologically assayed. Examples of products which are tested biologically at the laboratory are digitalis preparations and hormones.
Preparations including antibiotics are tested in the National Bacteriological Laboratory. The same applies to sterility tests of compounds intended for injection.
Preparations containing vitamins are tested in the vitamin department of the National Institute of Public Health.
A further measure in the control of the composition of a speciality is the requirement that the actual declaration of the contents of the drug be written in a standardized form, according to the Swedish Pharmacopoeia or national or international specifications. This implies, amongst other things, the use of generic names instead of proprietary names or codified chemical names for substances not yet assigned an international non-proprietary name.
A moderate variation of the declared content is usually allowed. In the case of a significant departure from the declared content, the manufacturer may be told of the results, and an explanation is sought before a final decision is reached. This means that several causes are investigated as a possible reason for the difference - e.g., deterioration of the product with time or various methods of assay.
Usual causes for rejection of the registration are departures from the declared content not satisfactorily explained or the finding of substances or other products not accounted for in the declaration.
Pharmaceutical Suitability
The quality of the product is thoroughly tested from a pharmaceutical point of view, as a rule according to the standards and the criteria of the Swedish Pharmacopoeia. The product's rate of deterioration is considered as specially important, and in many cases special tests are made to check this rate.
Important causes for rejection on grounds of pharmaceutical suitability are a high rate of deterioration or a large variation in the final product.
Medical or Therapeutic Usefulness
The experimental data on the action of the product or its active constituents in animals and man are thoroughly evaluated. Claims to an activity greater than that of other products are judged on the basis of comparative experiments.
Data supplied on acute toxicity (LD 50) as well as chronic toxicity tests in the case of products intended for prolonged use are considered, and toxicity data in various species and on similar substances are compared.
The medical and therapeutical usefulness is evaluated primarily by studying the available literature on the preparation as Well as the results of experimental and clinical tests not yet published.
The clinical data are evaluated and stress is laid upon those clinical trials that are designed to prove the " pure " or " net " action of the product as differentiated from other effects (hospitalization, psychological measures) or other treatment given concomitantly.
If a product has been used for a number of years in other countries and a comprehensive and adequate body of experiments and clinical experience is available, a review of the pertinent literature is usually sufficient for assessing the medical usefulness of a drug.
If the documentation is considered insufficient, however, as a first step the manufacturer or his representative is requested to present further support for the stated indications before a decision is reached. Sometimes a manufacturer is advised to arrange a controlled, statistically designed, clinical trial with the purpose of assessing the value of the product. Such a trial should if possible be organized as a double-blind study using control and test subjects with placebo administration. In the case of new products intended for use in a disease where specific treatment is already available, a double-blind study is suggested, using the best existing available specific treatment in the control group and the new drug to be studied in the test group.
Stress is also placed on the kind and frequency of side-actions and possible toxic reactions. Among these may be mentioned allergic symptoms, undue toxicity with regard to liver and kidney functions, the appearance of harmful disturbances in the hemopoietic system - e.g., methemoglo-binemia or agranulocytosis - or toxic reactions from other organs - e.g., from the gastrointestinal tract (hyperacidity, bleeding). Effects on the central nervous system - e.g., fatigue, drowsiness or sleepiness in the case of antiallergical preparations intended for day-use - or other symptoms are also considered.
Consideration is given not only to clinical manifestations of the drug in question, but also to known toxic actions of similar substances with parent chemical structure - e.g., possible liver toxicity by phenothiazine derivatives or bone marrow damage by amidopyrine compounds.
With regard to new substances with strong analgesic or antitussive action, special attention is paid to the possible addiction liability of such substances. Special attention is also paid to substances with stimulating properties and likely to be abused. Sweden complies with the international treaties in this respect, but may also declare substances which are abused to a high extent, but are not included among those internationally declared as addiction-producing drugs, as narcotic drugs and submit them to the same measures as are laid down for narcotics. This is the case with, for example, acetyloxiphenyl-methyl piperidine, amphetamine, dexamphe-tamine, metamphetamine, methylphenidate, phenmetraline and pipradol. These drugs are mainly used therapeutically for obesity or in certain nervous or mental states, but are beginning to be abused to a growing extent by juveniles and narcotic addicts.
The final step in the evaluation of the medical and therapeutic usefulness is to consider the indications and contra-indications actually claimed for the clinical use of the product intended for sale.
The usual causes of rejection of a preparation for therapeutic reasons are unjustified claims as to the clinical effect which the manufacturer is unwilling to withdraw, or a high incidence of untoward side-actions.
Price
Questions regarding price are handled by the financial section of the pharmacy division of the National Board of Health working in close co-operation. The price of the product is examined on the basis of the cost of the raw products, primarily taking into consideration the reasonable cost of production of the new compound. Other grounds for consideration are a comparison with the price of similar products which can be made as ex tempore preparations or which already exist as standardized preparations.
Advertising
The advertising material appended to the application for registration is examined with regard to the statements concerning indications, contra-indications and possible side-actions. Claims as to a favourable clinical effect, based upon reports comprising a few cases only and without controls, are considered as not justified, and have to be withdrawn if the product is to be sold.
When the investigation has been completed at the National Drug Control Laboratory, the bacteriological laboratory and the vitamin laboratory, and the price of the product has been established by the financial section of the pharmacy division, the results are presented to the Council on Drug Acceptance.
The Council on Drug Acceptance makes its recommendations based on the information gathered by the control agencies mentioned, and on experience and studies available from other sources.
The Council first decides on the basic question of whether the application for a permanent sales permit (registration) of a new product shall be granted or not. If the council recommends that registration be granted, the National Board of Health has the choice of following the recommendation or not. If the council recommends the rejection of an application, the National Board of Health is bound to concur.
If the information available is scanty or it is considered too early to assess the final value of a product, the council may defer its decision and may propose that the drug be sold on an individual sales permit or on a general ad interim sales permit (see section E), until more information on the use of the drug has been collected by doctors, hospitals, etc. In doubtful cases the council may propose further steps to be taken by various agencies before a final recommendation is made. In some cases the council may suggest specific studies to be carried out, recommending that the manufacturer or his representative induce interested hospitals or research workers to carry out a controlled clinical trial, based on a statistically designed plan with adequate controls.
In questions of major and fundamental interest the council may suggest an impartial study, designed as a statistically controlled clinical trial to be carried out by a special therapeutic trials committee appointed by the National Board of Health.
As examples of such investigations, mention might be made of three series of studies on the efficacy of chemotherapy in pulmonary tuberculosis, assessing the value of para-amino-salicylic acid (PAS), streptomycin and isonicotinohydrazide (INH). The first study in this series, carried out in 1948, consisted of a comparison between the effect of PAS in the test group and hospital treatment plus a placebo in the control group, because at that time no specific treatment was available. During the next study, carried out in 1950, placebo treatment could not be instituted, and the effect of PAS in the test group was compared to that of streptomycin in the control group. The third study in 1952 on the efficacy of INH was organized first as a comparison between INH in the test group and streptomycin in the control group, and later as a comparison between INH plus PAS in the test group and streptomycin plus PAS in the control group.
Other studies of this kind carried out by the therapeutic trials committee comprise an investigation of the use of antihistamines in the common cold [ 13] , and a study on the efficacy and possible side-actions of phenylbutazone in rheumatoid arthritis [ 14] , [ 15] , [ 16] .
The final decision on the acceptance or the rejection of a new drug for sale is made by the National Board of Health, based on the advice given by the Council on Drug Acceptance.
When the Board of Health accepts the drug it is included in the register of pharmaceutical specialities and given a registration number. This means a permanent sales permit as specified in the royal ordinance concerning trade in pharmaceutical specialities (1934).
If the application is rejected, the rejection is published and included in a separate list comprising the pharmaceutical specialities that are not to be sold.
The investigation of new drugs by the control authorities is very thorough and may require a long time. Procedures have therefore been introduced to make it possible to introduce pharmaceutical specialities on the market before the product is finally registered. Two such ad interim sales permits exist, an individual, temporary ad interim sales permit, and a general ad interim sales permit.
The Individual Temporary ad interim Sales Permit (licence)
Individual ad interim sales permits licences) are issued by the National Board of Health. As a rule, a licence is granted without any delay if a doctor, a veterinary surgeon or a dentist certifies that the preparation is to be used for scientific purposes only, or for a particular patient who can be shown to be in need of the preparation for the treatment of a specified disease.
The licence applies to a particular pharmacy and for a definite quantity of a certain drug. The drug will be delivered only on a prescription from the doctor, veterinary surgeon or dentist who has certified its need.
The individual ad interim sales permits are issued primarily for four different kinds of drug:
Drugs no longer imported or produced, which the manufacturer has been permitted to withdraw from registration;
Proprietary products which have not been filed for registration;
New drugs recently filed for registration but not yet registered and not given a general ad interim sales permit; and
Certain biologically active new drugs, where the clinical experience is not yet adequate to judge whether the drug has real value or not, or where it has not been possible to assess the real frequency of possible side-actions.
The licence procedure permits the board to obtain more detailed information about the drug than if the drug were to be sold freely. This information may come from the doctors that are using a new drug, and concerns the kind of disease treated, the indications and contra-indications for use and the number of patients actually obtaining the drug. Licensing was used for this purpose to a great extent during the years 1944 to 1948, when there was a limited supply of antibiotics and the use had to be restricted to patients really in need of these drugs - mainly penicillin and streptomycin.
The licence procedure can also be applied with regard to new analgesics or antitussives, the possible addiction liability of which has not been ascertained, and with regard to stimulating substances or other agents - e.g., those used for weight-reducing purposes - which are likely to be abused. This means that the number of doctors prescribing a certain drug, the number of patients getting it and the indications on which the drug is given can be controlled efficiently. If the drug were immediately released for sale, control measures would have to come afterwards - and in some cases too late.
Keeping a drug on licence while registration is pending has still another advantage. It makes it easier to induce the manufacturer to withdraw unjustified claims than if the preparation were already registered, because no licence will be issued until the manufacturer has complied with the existing rules.
The number of individual licences applied for in 1958 was 8,214, and in 1959, 9,798. The licences concerned about 800 different drugs; of these, 400 concerned drugs not yet being filed for registration, about 200 concerned drugs where registration was revoked (see section F) and 200 concerned drugs where application for registration was pending.
The usual action while a preparation is under consideration for registration is to issue a general ad interim sales permit for the product as early as possible. The permit is given, if no severe objections have been raised against the introduction of the drug on the market, at a preliminary consideration in the National Board of Health, in the National Drug Control Laboratory and in the financial section of the pharmacy division.
The general sales permit denotes that the sale of the preparation is not restricted to a particular doctor or pharmacy or patient, as is the case with the individual ad interim sales permit. All those drugs, for which a general ad interim sales permit has been granted, may be sold in every pharmacy according to the general regulations for the selling of drugs, may be advertised in medical journals and they may be handled in several other respects as if they already had been registered.
The general ad interim sales permit is issued by the National Board of Health by entering the preparation on the so-called licence-free list (licensfria listan) and including the name of the preparation in the quarterly list of pharmaceutical specialities accepted for sale without special licence (Apotekens förteckning över standardförpackade läkemedel) [ 8] . The product, however, is not given any registration number at this stage.
As a rule, in order to speed up proceedings and when the preliminary investigations of a drug disclose no grounds for serious criticism, the Council on Drug Acceptance is not consulted before a drug is placed on the licence-free list. Only in cases of doubt with regard to high frequency of side-actions, liability to produce addiction or abuse, or other specific grounds, are questions on the issuing of a general ad interim sales permit or individual temporary sales permits (licences) considered by the council.
The general ad interim sales permit as a rule remains in effect until the preparation is registered.
The drug may, however, be withdrawn from the licence-free list with immediate effect at any time by the National Board of Health. This implies that the general ad interim permit may be cancelled at any time and without the board having to consult the Council on Drug Acceptance. Such action may be necessary if, for example, during the further examination of the drug, factors should be found that necessitate immediate stopping of sale - e.g., undue toxicity, deviations from the declared content, unjustified claims in the advertising material, or too high a price.
The procedure of placing a preparation on the list of general ad interim sales permits (licensfria listan) has a high flexibility, and has proved very advantageous. Discriminate use of the licence-free list permits a good product which will be ultimately accepted to be sold shortly after the application for registration is filed, whereas a product not found fit for medical use can be rapidly withdrawn from the market.
On 1 January 1960 there were 2,488 registered specialities in Sweden and 353 drugs on the free list. The total number of drugs under application for registration at that time was 626. Thus, 57% of all new drugs were easily available on the market before the actual registration, the rest being available on licence only.
The final decision as regards registration may be delayed if it is considered advisable to obtain more data, especially regarding the clinical usefulness and the frequency of side-actions, before a product is definitely accepted. In this case the product is kept on licence or on the licence-free list, until a final decision can be made.
In accordance with the royal ordinance concerning trade in pharmaceutical specialities (1934), the National Board of Health, through its pharmacy inspectors, collects samples of the registered drugs on the market in order to ascertain whether they conform with the conditions under which they were accepted. Such market control is carried out routinely and usually with whole groups of products - e.g., hormones of various kinds, analgesics, chemotherapeutic agents, cardiac drugs, etc.
The control procedure may be instituted at any time for a certain drug if circumstances not apparent at the time of acceptance arise. Conditions which necessitate a market control may be an unexpected rapid deterioration of a preparation, an exaggerated or incorrect advertising campaign concerning the therapeutic value of the product or other laudatory remarks that might deceive the public, a sudden appearance of side effects from one or several batches of a product, a change in the composition of the product, etc.
The market control involves a partial or complete re-examination of the drug at the National Drug Control Laboratory including chemical, pharmaceutical and pharmacological rechecks and reassessment of the actual clinical value of the product in the light of the progress medicine may have achieved during the years which have elapsed since the introduction of the drug. The price is also reconsidered by the financial section.
If the re-examination during the market control shows that the registered product deviates from the original claims, the sale of the drug may be stopped by the National Board of Health by revocation of the registration. This means that the drug is cancelled from the specialities register - in urgent cases, immediately. Most commonly, however, the difference is reported to the manufacturer or his representative, and the manufacturer is given the chance of correcting the errors by his own efforts. If these fail or are considered inefficient, the question is referred to the Council on Drug Acceptance for consideration. After that, the registration may be cancelled by the National Board of Health.
The main difference between the revoking of the registration and the withdrawal of the drug from the licence-free list is that the control authorities can withdraw the drug from the licence-free list by immediate action, whereas the procedure for revoking the registration of a drug takes a certain time. This is one of the reasons that biologically active drugs may be kept on the licence-free list for a long time, as narcotics or drugs liable to be abused, if the clinical experience is insufficient for a final decision, or the frequency or severity of side-actions makes it likely that the clinical use of the drug may ultimately be restricted or not be allowed.
In conjunction with the examination of a new product by the National Drug Control Laboratory, the question also arises of whether the product shall be sold over the counter or on prescription only. The decision is based mainly, on the toxicity of the product, the indications and contra-indications for its clinical use, the frequency and kind of possible side-effects after therapeutic doses and after an overdose. The final decision is made by the National Board of Health.
The same procedure applies to the classification of a substance according to the royal statute on poisons.
Prescriptions for addiction-producing drugs under narcotic control cannot be re-used, and are filed at the pharmacies. This applies also to prescriptions for those drugs liable to be abused, which are placed under narcotic control by the Ministry of the Interior - viz., acetyloxiphenylmethyl piperidine, amphetamine, dexamphetamine, metamphetamine, methylphenidate, phenmetraline, pipradol. The authenticity of the narcotic prescription may be checked by the pharmacist by telephoning back to the doctor.
The narcotics prescriptions are collected, and can be subjected to control measures by the National Board of Health. Thus, it is possible for the board to check the number of prescriptions issued by a certain doctor, the drugs prescribed, the amount, and the names of the patients. Further, it is possible to check the prescription of a certain patient, how often the patient has seen a doctor, the number of prescriptions obtained, the kind of drugs, etc.
With regard to the medical profession, several lines are followed. Doctors are informed of new developments. Advertisements of narcotic drugs or drugs liable to be abused must contain a warning concerning the risks of narcotic addiction or drug abuse.
As regards individual doctors, several steps can be taken. The first measure by the National Board of Health is to issue a warning to a doctor who has not been cautious enough in prescribing narcotics. The next step is a partial restriction of the prescription rights; only one or two pharmacies in the vicinity of the doctor will be allowed to accept his prescription. The third step is a complete abolition of the right to prescribe narcotic drugs for either a short or long period. The final step is a revocation of the licence to practice medicine.
Close co-operation exists between the National Board of Health, the pharmacies and the police authorities. Criminal cases may include doctors prescribing more narcotics than is advisable from a medical point of view for themselves or for patients, or other individuals suspected of abusing narcotics by obtaining prescriptions for narcotic drugs from a number of doctors, or being involved in illegal drug traffic [ 17] .
Kungl. Maj: ts apoteksvarustadga den 14 november 1913, Svensk författningssamling, 1913: nr 308.
002Kungl. Maj: ts giftstadga den 26 november 1943, Svensk föfattnings-samling, 1943: nr 877.
003Kungl. Maj: ts kungörelse med vissa bestämmelser angående narkotiska ommen och beredningar den 16 september 1933, Svensk författningssamling, 1953: nr 559.
004Kungl. Maj: ts kungörelse angående kontroll och handel med vissa f'ör människor använda bakteriologiska preparat den 30 april 1925, Svensk författningssamling, 1925: nr 101.
005Kungl. Maj: ts kungörelse angående handel reed farmacevtiska specialiteter den 15 juni 1934, Svensk författningssamling, 1934: nr 306.
006LINDGREN, G.: Medicinen och reklamgranskningen - frihet under under ansvar, Den svenska marknaden, 1956: 12.
007- : Apotekens och industriens läkemedelsframställning i Sverige. Synpunkter på lagsfiftning och undervisning, Nordisk Medicin, 1953: 50.
008Apotekarsocietetens laboratorieavdelning: Apotekem register över standardförpackade läkemedel 1959.
009BACKMAN, S., BIRATH, G., BRUCE, 'Ir., GOLDBERG, L., KARTH, B., LEMMING, R.., LINDGREN, G. & LUNDQUIST, J.: "Para-aminosalicyclic acid treatment in pulmonary tuberculosis.", Amer. rev. tuberc, 1950: 61.
010ARDELL, T., EDFELDT, O., GOLDBERG, L., LEMMING, R., LEVIN, N., LINDGREN, G., LUNDQUIST, J. & NILSSON, O.: Para-aminosalicyclic acid (PAS) and streptomycin in pulmonary tuberculosis, Acta tuberc. scand., 1952: 27.
011ARDELL, R., BACKMAN, S., BRUCE, T., EDFELDT, O., GOLDBERG, L., GUSTAFSSON, B., LEMMING, R., LEVIN, N., LINDGREN, G., LUNDQUIST, J., NEUMÜLLER, H., NILSSON, O., NYRÉN, T., VON ROSEN, E., SELANDER, E. & STAHLE, I.: Traitement de la tuberculose pulmonaire par l'hydrazide de l'acide isonicotinique. Rapport préliminaire du Comité d'expérimentation thérapeutique de l'Association nationale suédoise contre la tuberculose. Etude portant sur 200 cas, Bull. un. int. tuberc., 1953: 23.
012- , - , - , - , - , - , - , - , - , - , - , - , - , - , - , - :
013GOLDBERG, L., HALLAN, L. & LINDGREN, G.: Unpublished observations.
014FJELLSTRA, K.-E., GOLDBERG, L., LINDGREN, G. & NILSSON, F: The therapeutic effect of phenylbutazone in active periods of rheumatoid arthritis. A control study, Int. congr. internal med., 3. Sthlm 1954. Proc., Sthlm 1956.
015- , - , - - , & NERI, A.: Phenylbutazone in active periods of rheumatoid arthritis. A methodological and controlled clinical trial. Acta med. scand., 1957, Suppl. 320.
016LINDGREN, G.: Phenylbutazon bei der Behandlung der aktiven rheumatischen Arthritis, Die Therapiewoche, 1958: 8.
017KROOK, G. & LINDGREN, G.: Kontrollen av handeln med narkotika i Sverige. En kommentar fill nu gällande bestämmelser, Sv. Läkartidn., 1954: 51.


