ABSTRACT
Introduction
Materials and methods
Cannabis leaf material
Results and discussion
Acknowledgements
Author: C. E. TURNER, C. Y. MA, M. H. RUSSELL, M. A. ELSOHLY
Pages: 43 to 54
Creation Date: 1981/01/01
A method for the determination of encapsulated d-limonene dimercaptan ( d-LDM), a possible marker for Cannabis sativa L. sprayed with herbicide, has been developed. The methodology includes a single-step extraction, followed by gas chromatography with a flame photometric detector (FPD) using n-octadecyl mercaptan ( n-octadecylthiol) as an internal standard. A linear relationship was obtained from spiked samples with an average coefficient of variation of 9.0 per cent. The method has been used to determine part-per-million levels of d-LDM in actual field experiments in Mexico. The FPD limit was found to be 7 ng for d-LDM.
Paraquat (1, l'-dimethyl-4, 4'-bipyridinium dichloride) is marketed worldwide and is the most widely used non-selective herbicide. In late 1975, paraquat was chosen as an agent for the chemical eradication of Cannabis sativa L. in Mexico [ 1] . Due to the fear of potential adverse and additive health effects caused by smoking marijuana contaminated with paraquat, the United States Department of Agriculture (USDA) began experimenting with chemical markers to be incorporated into paraquat solutions for use in Cannabis eradication programmes. The USDA selected d-limonene dimercaptan ( d-LDM) as a potential marker in these programmes. This compound is synthesized from naturally occurring d-limonene, a component of the oil of citrus peels, and hydrogen disulfide. It has an easily detectable and persistent foul odour and can be formulated and mixed for spraying along with any herbicide. The d-LDM used for field testing was prepared in microspheres by polymer encapsulation.l Encapsulation was designed to prevent the odour of d-LDM from dissipating between the time of application on Cannabis by spraying from a helicopter and the time of human consumption of crude marijuana products derived from that Cannabis. Subsequently, the polymer capsule was to erupt during smoking and release the foul odour of d-LDM, thus warning the user that a herbicide was present in the marijuana being smoked. In order to calculate the optimum amount of d-LDM needed per unit area of the field for odour detection, a method to quantify encapsulated d-LDM was needed.
The detection of trace quantities of sulfur-containing compounds in a complex matrix such as crude marijuana, extracts of marijuana, or marijuana smoke requires the elaboration of an extensive separation procedure. Alternatively, selective precipitating reagents such as silver nitrate [ 2] or a highly selective flame photometric detector (FPD) can be used to identify and quantitate d-LDM at the part-per-million level.
Since its introduction by Brody and Chaney [ 3] , the FPD has been widely employed for the selective detection and analysis of sulfur- and phosphorus- containing compounds such as are found in air-pollutant samples, coal-gasifier effluents, marine sediments, pesticide samples and tobacco smoke [4-8]. Although several limitations have been reported for this detector [9, 10], such as nonlinearity of its response, compound dependence for many classes of sulfur compounds and quenching of sulfur response in the presence of large hydrocarbon matrices eluting simultaneously with sulfur compounds, reliable quantification using gas chromatography (GC) with FPD can be achieved by careful optimization of the instrument and the selection of a suitable internal standard (IS).
A Hewlett-Packard gas chromatograph (5730A) interfaced with a flame photometric detector (18715A) set on sulfur mode (394 nm) was used throughout this study. Tests with d-LDM and thiram (bis(dimethylthiocarbamyl) disulfide) showed the optimal FPD response with a H 2 flow of 150 ml/min, O 2 flow of 20 ml/min and air flow of 100 ml/min. This combination gave a O 2/H 2 ratio of 0.27, in agreement with Burnett's observation [ 10] . The carrier gas (N 2) flow rate, which has been reported to be less critical in the overall FPD sensitivity [ 10] , was set conveniently at 60 ml/min. The column temperature (2% OV-17 on Gas Chrom Q, 100/120 mesh, packed in a 6 ft (1.8 m) x 2 mm ID silanized glass column) was set at 180°C for d-LDM and 200°C for thiram. Detector and injector temperatures were set at 200°C and 250°C, respectively. Under optimal conditions, the FPD limit was found to be 7 ng for d-LDM, when the signal-to-noise ratio was a minimum of 4:1.
Theoretically, the FPD response to sulfur compounds is proportional to the square of the sulfur mass-flow rate; but in many cases, the detector response has been reported to vary significantly, depending on factors such as
marker for Cannabis sprayed with paraquat 45 the design of the burner, the flame gas-flow rates and the type of compound being analysed [9-13]. However, by selecting a suitable internal standard, many of these variables can be eliminated. Thus, various classes of sulfur-containing compounds were evaluated as a potential internal standard. These included compounds containing dithiol functional groups, substances containing thiol or hydroxyl groups, and sulfur-containing compounds of approximately the same molecular weight as d-LDM. In all, a total of 16 compounds were tested; they are listed in table 1. Most of these compounds were eliminated because they eluted earlier than d-LDM, failed to extract from the spiked leaf samples with d-LDM (i.e., polarity difference), or decomposed in the presence of the components extracted from the Cannabis leaf, presumably due to oxidation, thus causing a nonlinear recovery rate from the spiked leaf sample. The compound n-octadecyl mercaptan ( n-octadecylthiol) was chosen as the internal standard because it exhibited none of these problems.
|
Dithiol compounds |
Compounds with thiol or hydroxy groups |
Compounds of approximately the same molecular weight as d-LDM |
|---|---|---|
|
1,9-Nonanedithiol
|
2-Aminothiophenol
|
Thioacetanilide
|
|
Hexadithiol
|
n-Octylthioethanol
|
Thioacetamide
|
|
3,4-Dimercaptotoluene
|
n-Octadecylthiol
|
2-Aminothiophene
|
|
Durene-
a,
a'-dithiol
|
Hexadecylthiol
|
p-Dithiane
|
|
Dithioerythritol
|
t-Tetradecylthiol
|
|
|
1,4-Butanedithiol
|
||
|
1,6-Hexanedithiol
|
All solvents used in the study were of reagent grade. A d-LDM technical sample (93% pure), unencapsulated, was obtained through the courtesy of Dr. W. A. Gentner of the USDA and was confirmed by GC/MS analysis as follows: m/e (per cent intensity) = 204 (1.0), 170 (2.6), 155 (1.3), 135 (1.6), 123 (3.2), 95 (2.6), 81 (3.5). The n-octadecyl mercaptan (b.p. 204-210°C/11 mm) used as an internal standard was purchased and used without further purification.
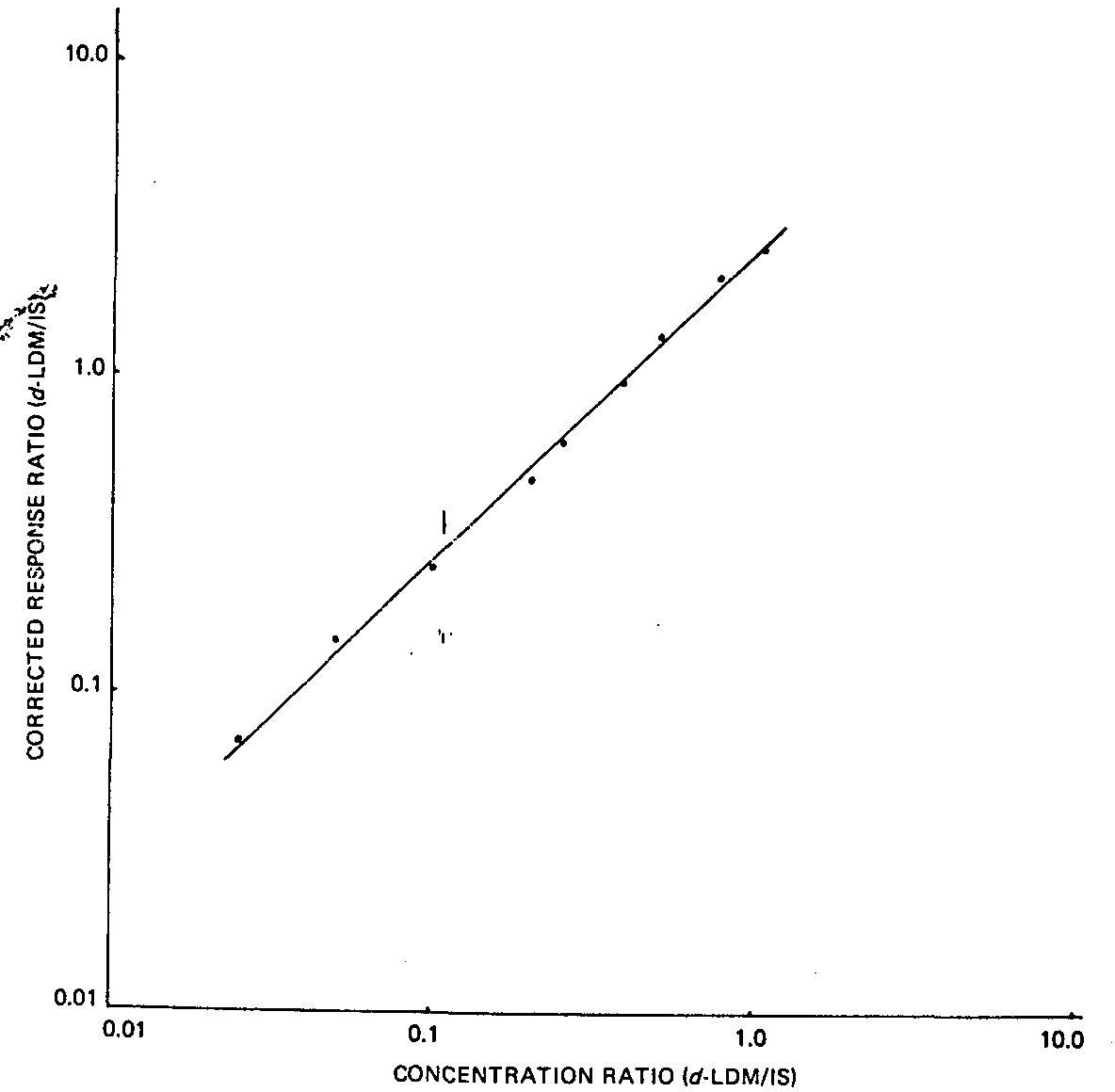
In order to determine the detector response to d-LDM, a suitable amount of d-LDM was added to 1 ml hexane solution containing 200 μg internal standard, so that the ratio of their concentrations ranged from 0.025 to 1.00. The resulting solutions were then analysed by GC/FPD. The ratio of the corrected response for d-LDM peak area to the corrected response for internal standard peak area versus the ratio of their concentrations was plotted using a log-log scale. The response factor was determined by linear regression analysis (correlation coefficient = 0.99) and could be expressed as log (ratio of corrected response) = 0.98 ? log (concentration ratio) + 0.406.
The corrected response, which was proposed by Maruyama and Kakemota (8), is peak area = √ H. W = CM where
H = peak height
W = peak width at half peak height
C = proportionality constant depending on the experimental conditions
M = mass of the sulfur
A graph of the data is in figure I.
A comparative study of detector response for d-LDM and the internal standard was also carried out. It was determined that the exponential proportionality constant for d-LDM was 2.39 and for n-octadecyl mercaptan was 2.21. The correlation coefficients of linear regression analysis were found to be 0.99 for both compounds (see figure II). Thus it was concluded that the two compounds behaved similarly with respect to FPD and approximately followed the square law.
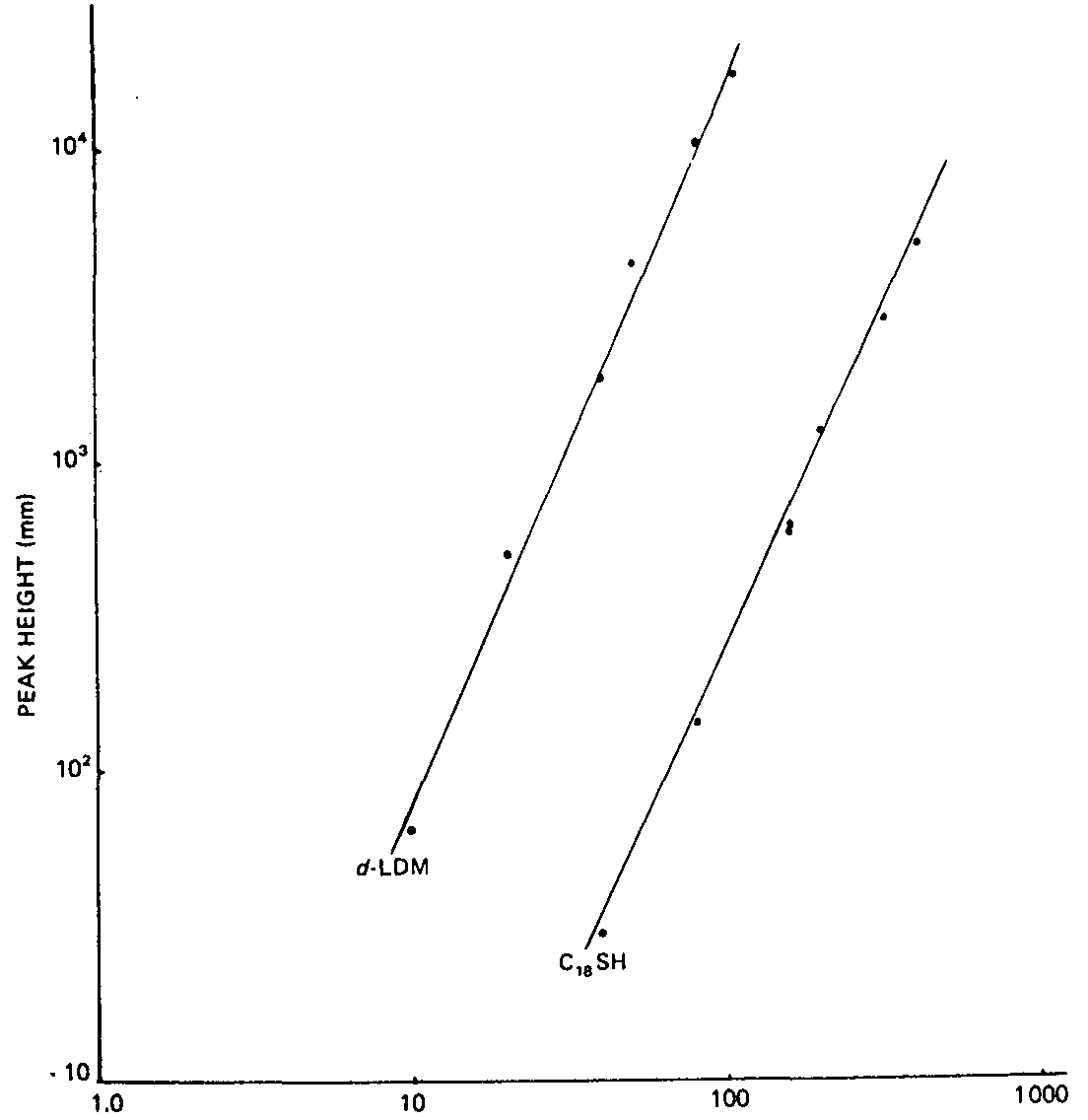
SAMPLE SIZE (ng)
All d-LDM-free Cannabis leaf samples were female Mexican ( Cannabis sativa L.) grown in Mississippi from known seed stock. All leaf material was air-dried and passed through a 12-mesh sieve prior to weighing.
Three samples of Cannabis leaves were obtained from three different fields in Mexico. Each field was sprayed with a mixture of paraquat and micro-encapsulated d-LDM. The solution was formulated to deliver 4% d-LDM on the plants with each helicopter spray pass. The paraquat solution was the stock material used in the Mexican eradication programme.
Since neither hexane nor chloroform extracts of dried Cannabis leaf samples showed any detectable response on FPD at the column temperature of 180°C, these two solvents were used to evaluate the extraction efficiency. A typical extraction was carried out as follows:
To 1 g of dried leaf sample of field-sprayed Cannabis were added 20 μl of solution containing 10 mg/ml internal standard in hexane. Also d-LDM in quantities ranging from 10 to 150 μg were added to the d-LDM-free samples for use in the construction of calibration curves and for the determination of recovery rates;
Each leaf sample was then sonicated in 40 ml of hexane (or chloroform) for 20 min. The mixture was then passed through a glass-wool plugged funnel to remove the marc;
After evaporating the solvent under vacuum at 35-40°C, the resulting residue was dissolved in 1 ml hexane and a 0.5 to 1.0 μ1 aliquot was injected into the gas chromatograph.
In order to estimate the absolute recovery rate of unencapsulated d-LDM from marijuana, extraction of triplicate 1-g leaf samples spiked with 50 ppm d-LDM [ 2] was carried out and 200 μg internal standard added prior to analysis by GC/FPD. Using the response factor determined as described above, the recovery rate was found to be 72 per cent. Although the recovery from the hexane extraction was slightly lower than that from chloroform, the residue produced from hexane extraction was less (6.3 weight per cent as compared to 7.6 from chloroform extraction). In the case of hexane, the moderate recovery rate obtained with hexane could possibly be attributed to interference from the low-molecular-weight hydrocarbons eluting near the retention time of d-LDM as illustrated using a GC/FID (flame ionization detector) system (figure III). Consequently, the FPD response for d-LDM was lower than that of the internal standard. With chloroform, on the other hand, many of these hydrocarbons will not be extracted; however, several cannabinoids will be extracted which interfere with the internal standard peak. Based on the extraction efficiency alone, hexane was chosen as the extraction solvent.
The corrected response rate of d-LDM versus internal standard obtained from the spiked sample at a given d-LDM/IS concentration ratio was consistently lower than that obtained from stock solution (for example, 0.49 from spiked sample as compared to 0.62 from stock solution with a d-LDM/IS concentration ratio of 0.25). Therefore, a calibration curve based on the observed corrected response ratio versus the d-LDM/IS concentration ratio in the spiked leaf samples was determined (figure IV). By linear regression (correlation coefficient = 0.99) and from the calibration curve, it followed that the ratio of corrected response = 0.0063 ? (ppm of d-LDM added) + 0.161. Each point on figure IV represents the mean of quadruple determinations. The average coefficient of variation over a period of three weeks was found to be 9.0 per cent.
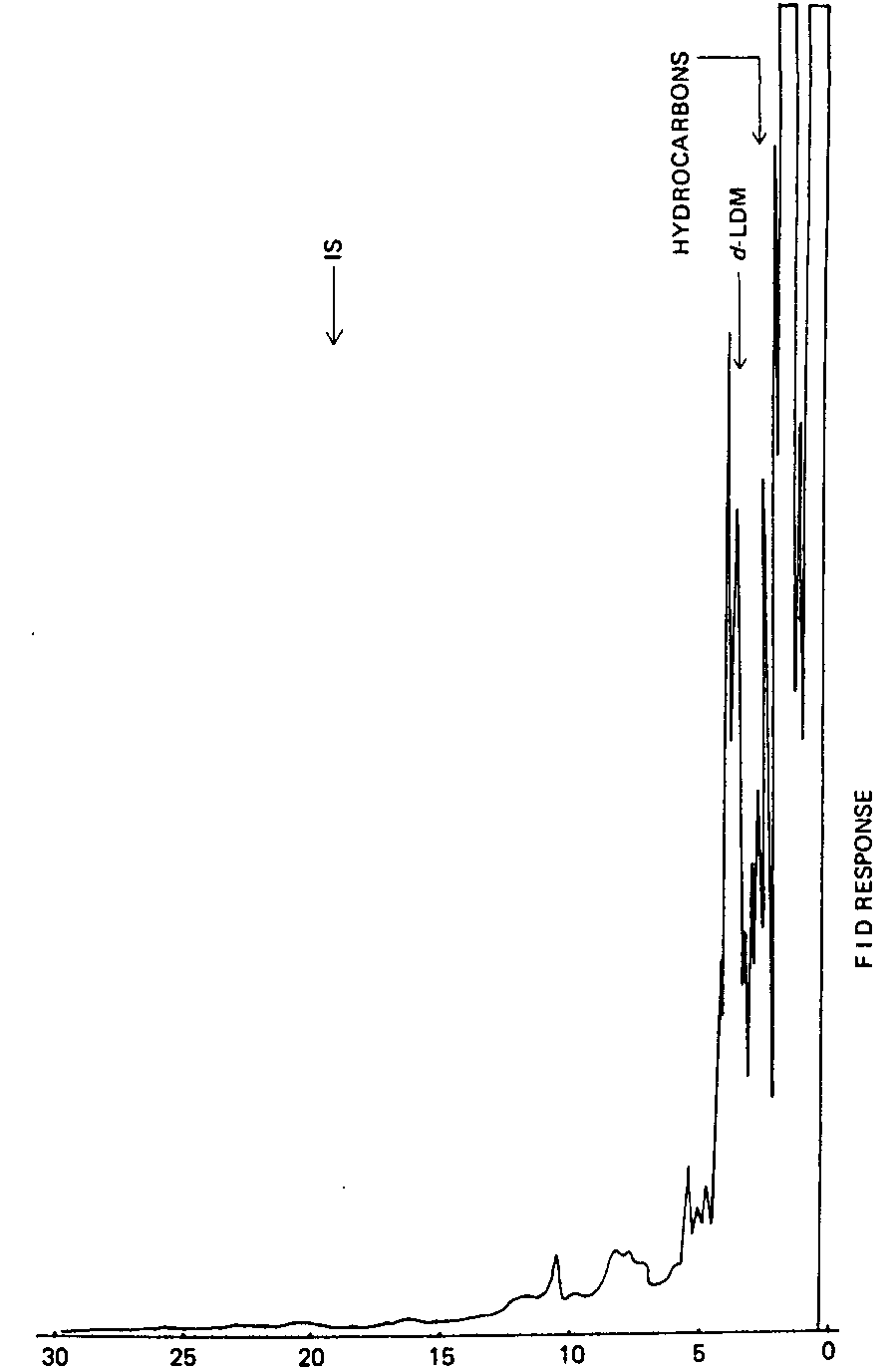
RETENTION TIME, IN MINUTES
Scanning electron microscope analyses of Cannabis samples from Mexico showed that very few spheres remained whole after spraying [ 14] . No obvious odour of d-LDM was present in any of the dried samples when they reached our laboratory. Moreover, no odour of d-LDM was obvious when the treated marijuana was slowly burned; this suggested that no encapsulated d-LDM was present or, if it were, the concentration was below the minimum required to overcome the natural odour of burning marijuana. If d-LDM were to be detected using the analytical procedure discussed, it would (a) verify that the relatively non-polar hexane could penetrate the encapsulating spheres and extract d-LDM, (b) provide some data on the relative amount of encapsulated d-LDM required to overcome the natural odour of burning marijuana, and (c) provide data on the possible application of this method to large-scale screening of samples for d-LDM in the future.
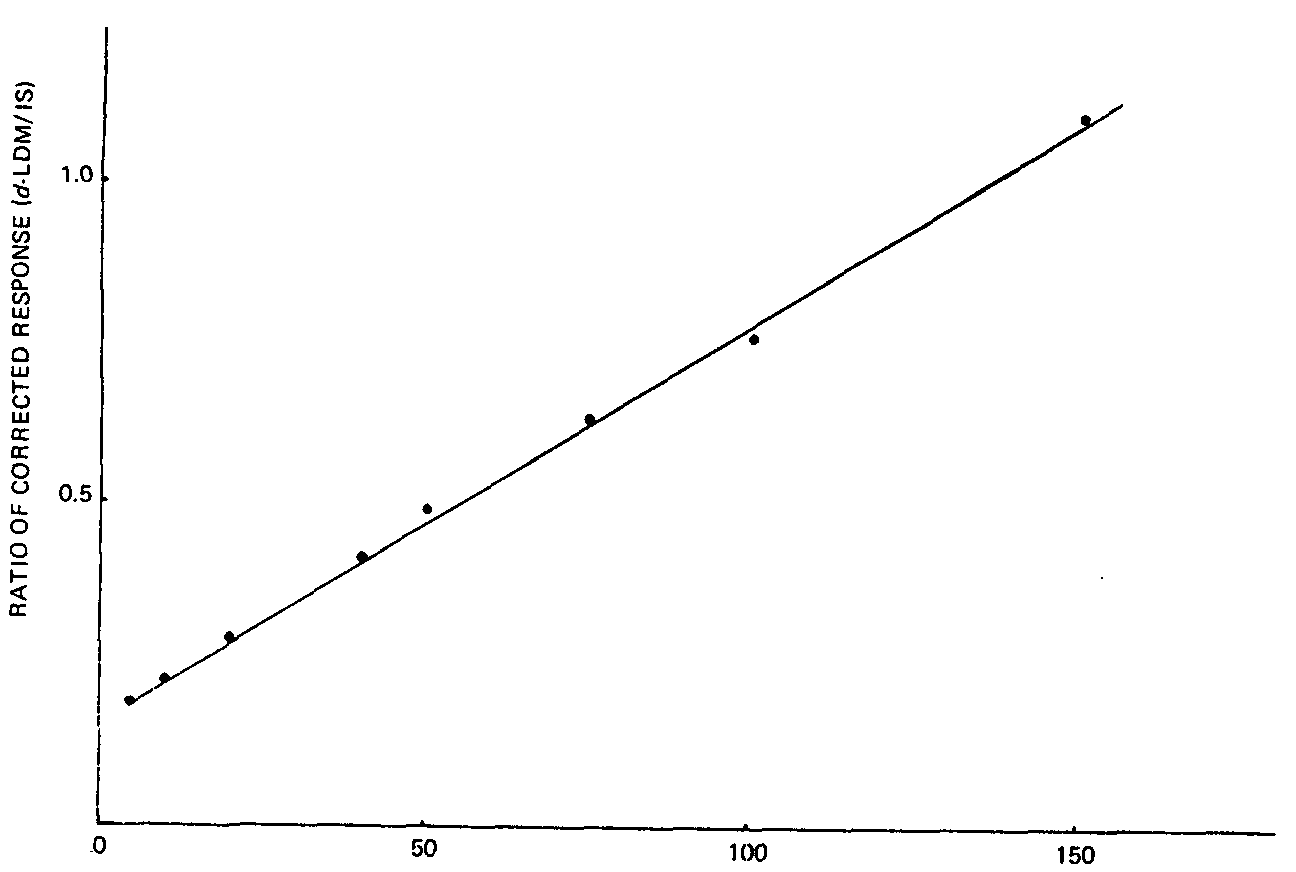
PARTS PER MILLION OF d-LDM ADDED
Since encapsulated d-LDM was formulated to provide 4 per cent of active d-LDM, one sample was selected from each of the three experimental plots in Mexico sprayed via helicopter. Sample No. 1 was collected from a field (field No. 3) sprayed twice, the routine procedure in Mexico. Sample No. 2 was from a field (field No. 5) sprayed three times and sample No. 3 was from a field (field No. 4) sprayed five times. All three fields were sprayed with the same solution on the same day from the same helicopter and pilot.
Theoretically, these fields should have received a d-LDM concentration of 8, 12 and 20 per cent, respectively. Also, theoretically, the paraquat concentrations received by the fields should follow a linear relationship. Using the procedure reported by Turner et al. [ 1] paraquat analyses were performed on samples No. 1, No. 2, and No. 3 which were collected at 24, 20 and 18 h after spraying, respectively. The data obtained from these analyses demonstrate that the theoretical concentrations calculated are very difficult to obtain under d-Limonene dimercaptan, a herbicide marker for Cannabis sprayed with paraquat 51 actual field conditions. As shown in table 2, field No. 4, sprayed five times, contained 1,080 ppm of paraquat; field No. 3, sprayed two times, contained 347 ppm; and field No. 5, sprayed three times, contained 286 ppm.
The relative ratios of 347 ppm for two sprayings and 1,080 for five sprayings are within acceptable values, taking into consideration factors such as variations in plant size, variable wind velocities, differences in number of hours after spraying that the samples were collected etc. The paraquat level in field No. 5 would be expected to have a value between those observed for fields No. 3 and No. 4 instead of the smaller concentration obtained (286 ppm). However, an examination of the actual physical conditions leads to a possible explanation. Field No. 5 was located on a river bank surrounded by many large trees, one of which was situated directly in the flight path of the helicopter. For safety reasons the pilot was forced to approach this field at an altitude higher than that used in spraying the other fields, thus preventing a continuous and uniform spray application. These facts, along with the fluctuation of plant size, random selection of the samples and size of the field created conditions that would greatly influence the amount of spray per unit area of leaf.
Since the d-LDM was incorporated into the paraquat solution, a wide fluctuation would also be expected in the concentrations of d-LDM found in the respective samples. Using the analytical method described above, the concentrations of d-LDM detected for fields No. 3, No. 4 and No. 5 were found to be 86.9 ± 8.3, 136.7 ± 11.6 and 27.3 ± 1.6 ppm (table 2); assuming the extraction process affords a 72 per cent recovery rate for d-LDM, these concentrations would be 120.6 ± 8.3, 189.9 ± 11.6 and 37.9 ± 1.6 ppm. These data fit relatively well with the paraquat data. The ratio of d-LDM to paraquat (table 2) varies from 9.5 to 25.0 per cent and does not fit the expected pattern. However, the proportion of disintegrated encapsulating spheres has not been determined; thus, even under rigidly controlled conditions these data might not fit a pattern.
Note: Samples were collected on 5 June 1979 and analysed on 4 October 1979.
aDetermined by methods reported in [1].
bBased on 4 per cent per spray application.
cBased on actual detectable amounts. No corrections made to reflect extraction efficiency or recovery rate.
The data in table 2 validate the procedure described for extracting d-LDM from the encapsulating sphere. From the data obtained and assuming a 72 per cent recovery rate, it is obvious that more than 189.9 ± 11.6 ppm of encapsulated d-LDM are needed to overcome the natural odour of burning marijuana. With the minimum amount detected being below 50 ppm and the detector limit being 7 ng, or 7 ppb for a 1-g sample, this procedure is sensitive enough to be very useful in any screening programme should d-LDM be used in volume as a herbicide marker (figure V).
Cannabinoid profiles of the samples are shown in table 3. Literature reports on the fluctuation of cannabinoids with age are numerous [ 15] [ 16] . Thus, with these samples being collected at different stages of growth, no correlation of the cannabinoid data would be expected. Cannabinoid assays were determined using the procedure described by Turner's group [ 17] [ 18] .
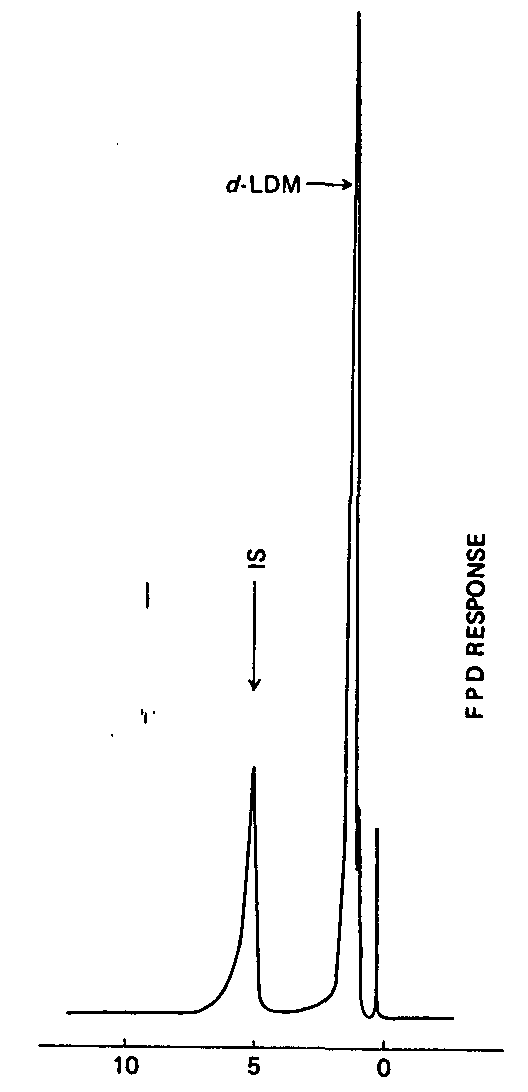
RETENTION TIME, IN MINUTES
Notes:
1. CBDV = cannabidivarin; Δ [ 9] -THCV = (-)-Δ [ 9] - trans-tetrahydrocannabivarin; CBL = cannabicyclol; CBD = cannabidiol; CBC = cannabichromene; CBGM = cannabigerol monomethyl ether; Δ [ 8] -THC = (-)-Δ [ 8] - trans-tetrahydrocannabinol; Δ [ 9] -THC = (-)Δ [ 9] - trans-tetrahydro-cannabinol; CBG = cannabigerol; CBN = cannabinol.
2. t = trace.
The authors gratefully thank Dr. O. J. Bouwsma for obtaining mass spectral data and Ms. Lolita Torres for obtaining paraquat analyses. Special thanks are due to Dr. Alan B. Jones, to Dr. Walt Gentner of the USDA, to Lic. Oscar Flores, the Procurador General de la República Mexicana, to Mr. Sergio Zapata and other members of the Procurador General's staff. This research was supported by the Research Institute of Pharmaceutical Sciences and in part by NIDA contract # 271-78-3527.
1Micro-encapsulated d-LDM was prepared by Pennwalt Chemical Co. at a concentration of 15.2%.
2Technical grade d-LDM. See above under the heading Chemicals.
C. E. Turner and others, "Detection and analysis of paraquat in confiscated marihuana samples", Bulletin on Narcotics , vol. 30, No. 4 (1978), pp. 47-56.
002J. S. Marheuka and S. Siggia, "Atomic absorption method for determining micromolar quantities of thiols", Analytical Chemistry, vol. 51, 1979, pp. 1259-1262.
003S. S. Brody and J. E. Chaney, "Flame photometric detector: the application of a specific detector to phosphorus and sulfur compounds sensitive to subnanogram quantities", Journal of Gas Chromatography , vol. 4, 1966, pp. 42-46.
004R. K. Stevens and others, "Gas chromatography of reactive sulfur gases in air at parts-per-billion level", Analytical Chemistry, vo1.43, 1971, pp. 827-831.
005S. K. Gangwal and D. E. Wagoner, "Response correlation of low molecular weight sulfur compounds using a novel flame photometric detector", Journal of Chromatographic Science , vol. 17, 1979, pp. 196-201.
006T. S. Bates and R. Carpenter, "Determination of organosulfur compounds extracted from marine sediments". Analytical Chemistry, vol. 51, 1979, pp. 551-554.
007C. G. Daugton. A. M. Cook and M.. Alexander, "Gas chromatographic Determination of phosphorus-containing pesticide metabolites via benzylation", Analytical Chemistry vol. 51, 1979, pp. 1949-1953.
008L. Biomberg, "Gas chromatographic separation of some sulfur compounds on glass capillary columns using flame photometric detection", Journal of Chromatography , vol. 125, 1976, pp. 389-397.
009M. Maruyama and M. Kakemota, "Behaviour of organic sulfur compounds in flame photometric detectors", Journal of Chromatographic Science , vol. 16, 1978, pp. 1-7.
010C. H. Burnett, D. F. Adams and S. O. Farwell, "Relative FPD responses for a systematic group of sulfur-containing compounds", Journal of Chromatographic Science , vol. 16, 1978, pp. 68-73.
011C. H. Burnett, D. F. Adams and S. O. Farwell, "Potential error in linearized FPD responses for sulfur", Journal of Chromatographic Science, vol. 15, 1977, pp. 230-233.
012J. G. Eckhardt, M. B. Denton and J. L. Moyers, "Sulfur FPD flow optimization and response normalization with a variable exponential function device", Journal of Chromatographic Science , vol. 13, 1975, pp. 133-138.
013P. L. Patterson, F. L. Howe and A. Abu-Shumays, "Dual-flame photometric detector for sulfur and phosphorus compounds in gas chromatograph effluents", Analytical Chemistry , vol. 50, 1978, pp. 339-344.
014L. T. Harmison, Mexican Field Tests of Marihuana Marker (Washington, D.C., Department of Health, Education and Welfare, Public Health Service, 1979).
015C. E. Turner and others, "Constituents of Cannabis sativa L. XV: Botanical and chemical profile of Indian variants", Planta Medica , vol. 37, 1979, pp. 217-225.
016C. E. Turner and others, "Constituents of Cannabis sativa L. XIV: Intrinsic problems in classifying Cannabis based on a single cannabinoid analysis", Journal of Natural Products , vol. 42, No. 3 (1979), pp. 317-319.
017C. E. Turner and K. Hadley, "Constituents of Cannabis sativa L. II: Absence of cannabidiol in an African variant", Journal of Pharmaceutical Sciences , vol. 62, No. 7 (1973), p. 1083.
018C. E. Turner, K. W. Hadley and K. H. Davis, "Constituents of Cannabis sativa L.V: Stability of an analytical sample extracted with chloroform", Acta Pharmaceutica Jugoslavica , vol. 23, No. 2 (1973), p. 89.


