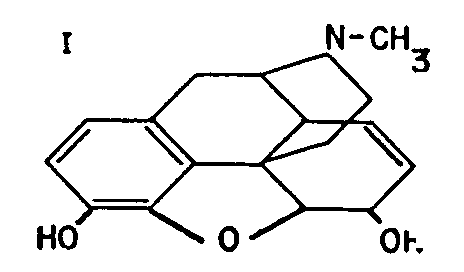
I
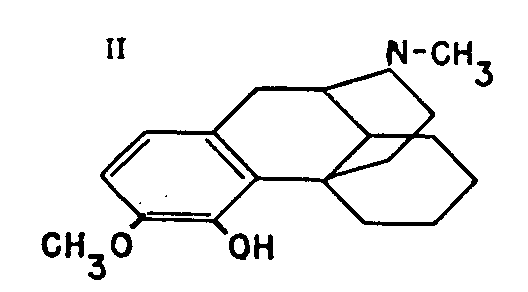
II
Morphinane Series
The Pethidine Series
Methadone Series
Other Series of Completely Synthetic Analgesics
NOTES
Author: Paul B. Weill, Ulrich Weiss
Pages: 12 to 31
Creation Date: 1951/01/01
The history of chemical research on morphine and the alkaloids chemically related to it has been described in a previous article in this Bulletin.*
The main arguments in favour of formula I, below, for morphine proposed by Gulland and Robinson 1** in 1925 were set out in that article, together with the objections to its unanimous adoption. The article ended with the hope that the synthetic experiments now in progress would provide the irrefutable proof so long awaited. That hope has been very quickly fulfilled. In the remarkable research work referred to at the end of the previous article, Grewe et al[2] have confirmed Gulland and Robinson's formula by a total synthesis of tetrahydrodesoxycodeine (II), identical with the corresponding compound derived from codeine.
* United Nations Bulletin on Narcotics, Vol. II, No. 2, page 8.
** Superior figures refer to the bibliography at the end of this article.
An earlier article by Paul Weill has treated of the chemistry of the natural analgesics, morphine and codeine, and has especially discussed the work that led to the elucidation of their molecular structure. The object of the present article is the chemistry of artificial analgesics, discussing first the substances obtained by chemical transformation of the natural alkaloids, and subsequently the totally synthetic drugs. The first of those groups obviously retains the essential carbon-nitrogen skeleton of morphine; among the totally synthetic analgesics, one group also contains this skeleton, while others have only remote chemical analogies with morphine
3This second article will discuss the analgesics obtained either by transformation of natural alkaloids or entirely by synthesis. The first group of partially synthetic analgesics will be dealt with separately from the group of compounds more recently prepared by total synthesis.
Since the chemical research which produced the analgesic compounds discussed in this review was under-taken in the hope of discovering analgesics free from the undesirable secondary actions of morphine, it will be necessary to preface the chemical part of this article with a very brief outline of the principal pharmacological properties of morphine, with particular emphasis on those side-effects which constitute the most serious clinical drawbacks.
Morphine acts as an almost universal analgesic against pain in its various forms. When a large enough dose is administered, analgesia is usually followed by sleep.
This sequence is characteristic of morphine, whereas other types of medical preparations are either hypnotic but not analgesic (barbiturates) or antalgic but not hypnotic (salicylates).
The analgesic action of morphine is accompanied by numerous secondary effects, some of which are particularly objectionable. The worst of these are habituation and addiction, which increase rapidly with use. Much research has been inspired by the hope of separating those undesirable side-effects of morphine from its analgesic properties, but despite the large number of compounds prepared, only very limited success has been achieved. The recent discovery of synthetic products, a number of which have only a distant chemical relation to morphine, but which, like morphine, are both analgesic and habit-forming, would unfortunately appear to indicate that these properties are closely, and in all probability necessarily, inter-related.
The stimulation which morphine sometimes causes in human beings is regularly produced in certain animals like the cat. Thebaine produces this convulsive effect to such an extent even in man, that it may be regarded as a spastic poison, like strychnine. The respiratory centre is depressed by even small quantities of morphine, and the use of morphine in obstetrics is limited by the fact that minute doses produce this bulbar depression to a dangerous degree in the foetus and the new-born infant.
The action of morphine on the intestinal tract consists in lowering the tonus of the small intestine, and in causing constipation. In some cases it has a motor action on the stomach and a central emetic action. The latter property is predominant in the case of apomorphine.
Generally speaking, the morphine derivatives may be said to resemble morphine in their pharmacological properties. However, this analogy is only qualitative, since quantitatively they may differ considerably, not only with respect to the active doses, but also in the sequence and intensity of their effects. To the two examples already given, of thebaine and apomorphine, we shall add merely that of codeine, which is definitely inferior to morphine in its analgesic action, but which has no central depressive effect and is especially active as a cough sedative.
In this section, those analgesics shall be discussed which are obtained from the natural alkaloids; since it is hardly feasible to mention every one of the numerous compounds prepared so far, only the more important and representative substances are considered.
We shall include under this heading, the analgesics obtained from the natural morphine alkaloids, but as we believe it to be unnecessary to refer to each one of the many compounds which have so far been prepared, we have confined ourselves to the most representative and important of them. The order of presentation follows no rigid system, since it was felt that any classification, whether chemical or pharmacological, was artificial and unimportant. Although this study is confined in principle to their chemistry, it appears essential to give a brief description of the main pharmacological and clinical characteristics of the compounds under consideration.
This alkaloid, commonly known as heroine, is obtained by the acetylation of the two hydroxyls of morphine. It is typical of the derivatives obtained by esterification of both the phenolic and alcoholic functions of morphine with various acids (propionic, valeric, benzoic, etc.).
Although its analgesic action is stronger and faster than that of morphine, heroine has only limited use because of its great drawbacks and dangerous features. It is more toxic than morphine, and very rapidly produces habituation and addiction, accompanied by intense euphoria and excitation. These latter effects make the heroine addict particularly dangerous. The manufacture and use of heroine are prohibited in many countries, including the United States.
Codeine (III) is produced when the phenolic hydrogen of morphine is replaced by a methyl group. It is present in opium but in quantities insufficient to meet the requirements of therapeutic practice. It is therefore prepared by methylation of morphine and we are including it among the partially synthesized derivatives. Codeine may be regarded as the first of a series of phenolic ethers of morphine, the most important of which are ethylmorphine or dionine, and benzylmorphine or peronine.
Inferior to morphine in its analgesic properties, codeine is also less toxic, while some of its undesirable secondary actions, such as the danger of addiction and the effect on the intestine, are much less pronounced. Codeine is used as an analgesic where the pain is not too severe, and especially as a cough sedative, because of its non-depressive action on the respiratory centre, a feature which makes it preferable to morphine.
As an analgesic, dionine or ethylmorphine lies between morphine and codeine and is similar to the latter in its properties, particularly as a cough sedative. Its action is regarded as superior to that of codeine, but is accompanied by greater toxicity. Dionine is sometimes used in ophthalmology because of its power to produce irritation and hyperaemia of the eye, desirable in certain cases.
Benzylmorphine or peronine is a more active but also a more toxic substitute for codeine.
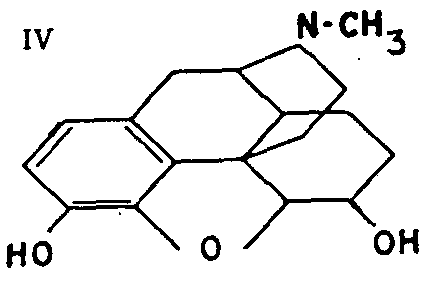
This compound (IV) results from the catalytic hydrogenation of the double bond of morphine in the presence of colloidal palladium. It is regarded as preferable to morphine, for which it is a substitute, by certain authorities who claim that it is a more powerful analgesic and less convulsive and toxic. It is not widely used, however; it has not been introduced in the United States and is hardly used any more in Europe.
Obtained by the hydrogenation of the double bond of codeine, this derivative has suffered the same fate as dihydromorphine.
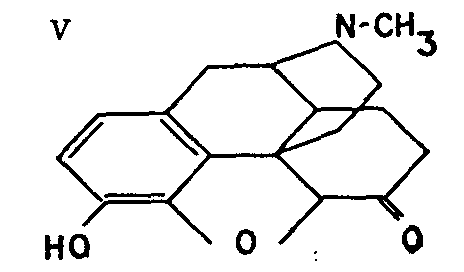
This compound (V), prepared by the Knoll Laboratories in 1926, results from the rearrangement of morphine under the action of catalysts like platinum or palladium, in the presence or absence of hydrogen.
Dilaudid has the advantage of being a more powerful analgesic than morphine, but this is counter-balanced by greater toxicity. While possessing the same general properties as morphine, dilaudid has a number of individual features which in some cases make it preferable to morphine: less habit-forming, according to some authorities; favourable action on post-operative intestinal spasms; less pronounced constipating and emetic action than morphine.
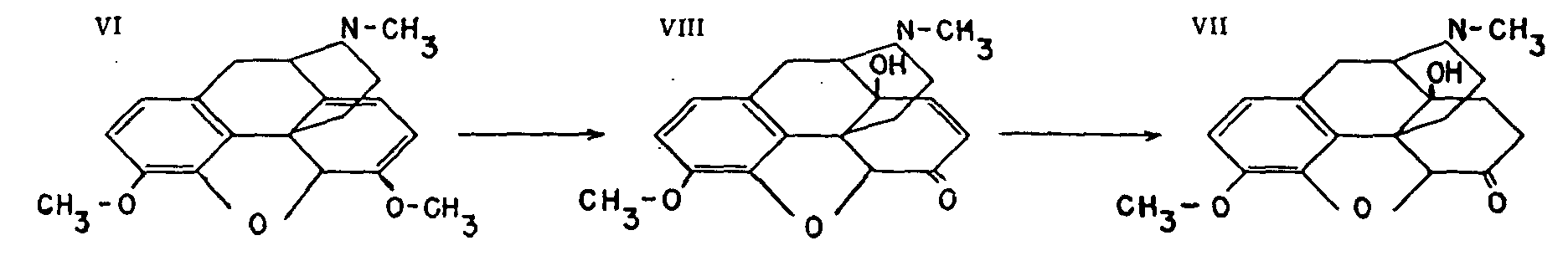
This methyl ether of dilaudid may be prepared from codeine or thebaine. Under the influence of the same catalysts which transform morphine into dilaudid, codeine is similarly rearranged to give dihydrocodeinone. Thebaine (VI) also gives the same compound by the acid hydrolysis of dihydrothebaine (XIII) ( see p. 15), its hydrogenation product.
A number of authorities ascribe to dihydrocodeinone properties which lie midway between those of morphine and codeine. Its most important feature is its activity as a cough sedative. The acetate of the enol form of dihydrocodeinone has been proposed in Germany as an antitussive under the name acedicon (XIV) ( see p. 15).
This compound (VII) is produced by the catalytic hydrogenation of hydroxycodeinone (VIII), which is itself obtained by the action of hydrogen peroxide on thebaine (VI).
Eucodal, a substitute for morphine, lies midway between codeine and morphine in its action and is recommended as an analgesic. Of German origin, it is not used in the United States.
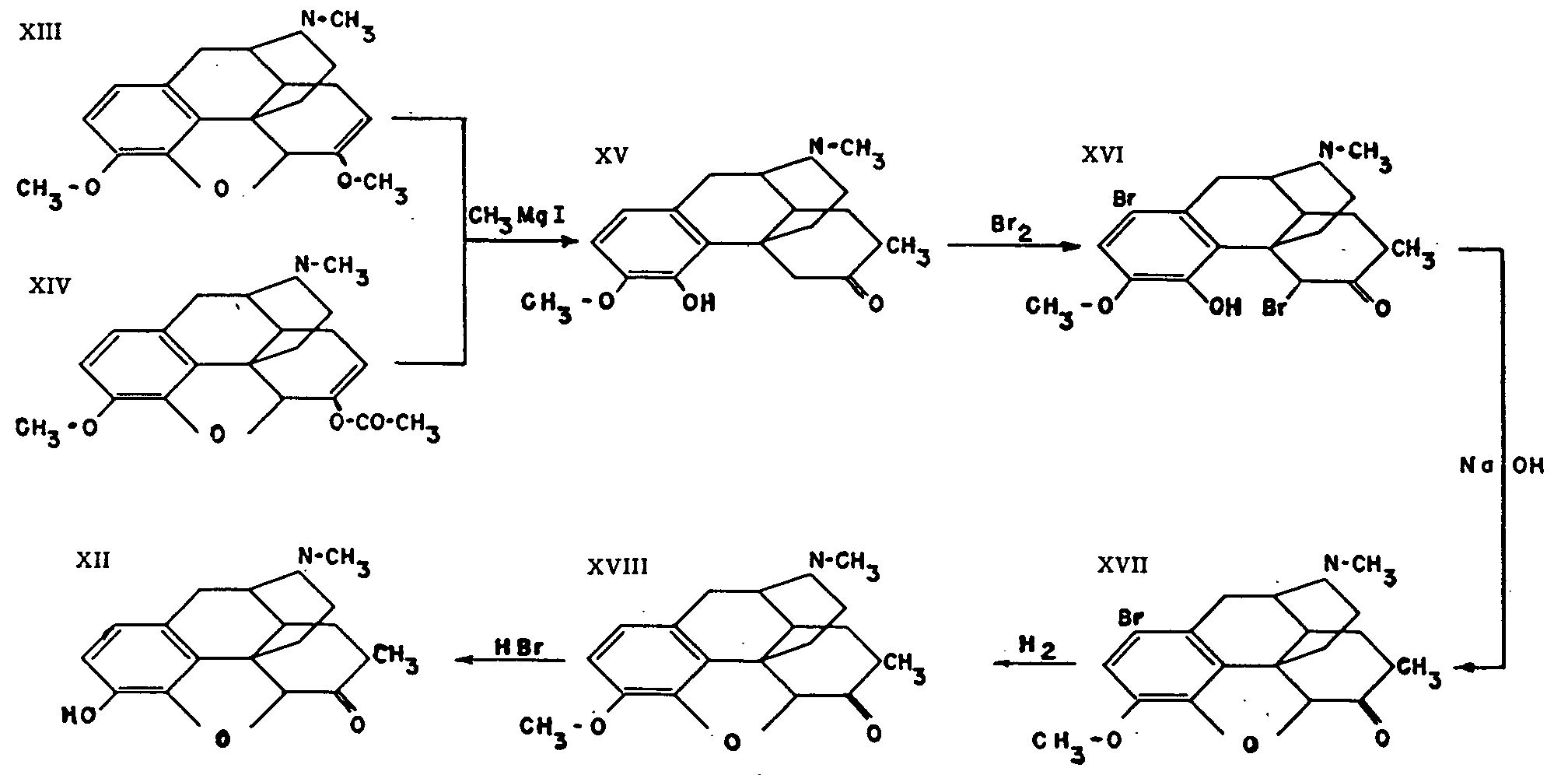
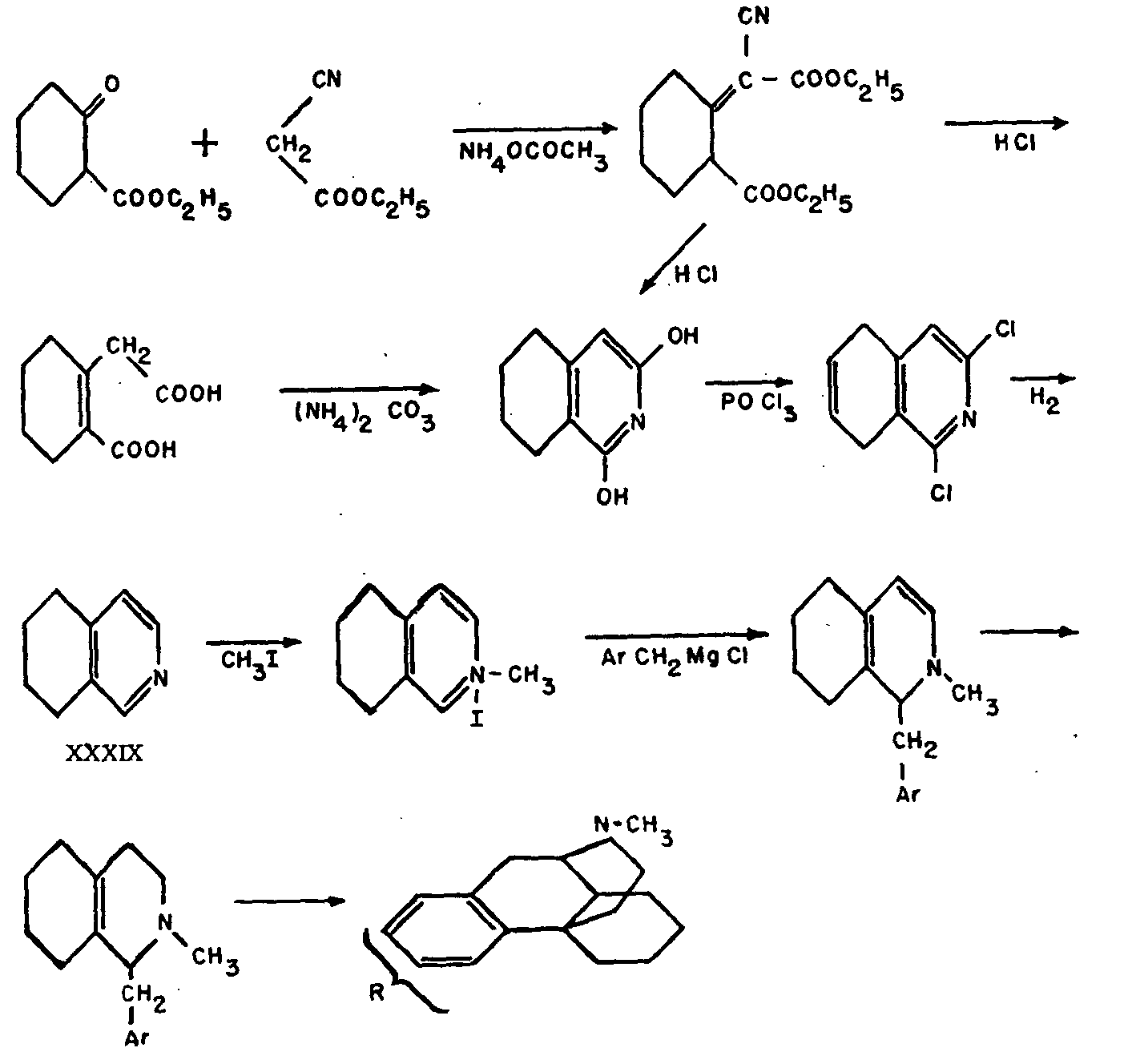
This compound (IX) was discovered by Small[3] in the course of his research on those derivatives of morphine and codeine in which oxygen is no longer attached to carbon atom 6. It is prepared from α-chlorocodide (X), which is itself obtained by the action of thionyl chloride on codeine (III). By catalytic reduction, α-chlorocodide (X) gives dihydrodesoxycodeine D (XI), which yields dihydrodesoxymorphine D on demethylation.
This substance appears to be remarkably active, with an analgesic effect about ten times greater than that of morphine; its toxicity, although likewise exceeding that of morphine, is apparently only three times as great. Its action is described as being very rapid but of short duration, and not accompanied by vomiting. It has been given the commercial name of permonid in Switzerland, where it is used.
The constitution of this compound (XII), which was likewise discovered by Small[4] , has not yet been established beyond doubt. This uncertainty springs from the first reaction in the synthesis of metopon, the action of methyl-magnesium halide on dihydrothebaine (XIII), which is difficult to interpret for reasons discussed in the previous article.

The successive stages in the synthesis of methyldihydromorphinone are represented in the accompanying diagram taken from a recent publication by Small[5] on this subject. It will be noted that the first reaction mentioned actually produces two isomers, the manner of formation and constitution of which still defy satisfactory explanation.
Metopon has been studied clinically only in cases of inoperable cancer. Its action has been ascertained by oral administration and it has been found to have few undesirable side-effects. The addiction to which it gives rise is less pronounced than that caused by morphine. Despite these promising indications, it would be premature to form any final judgment as to its value.
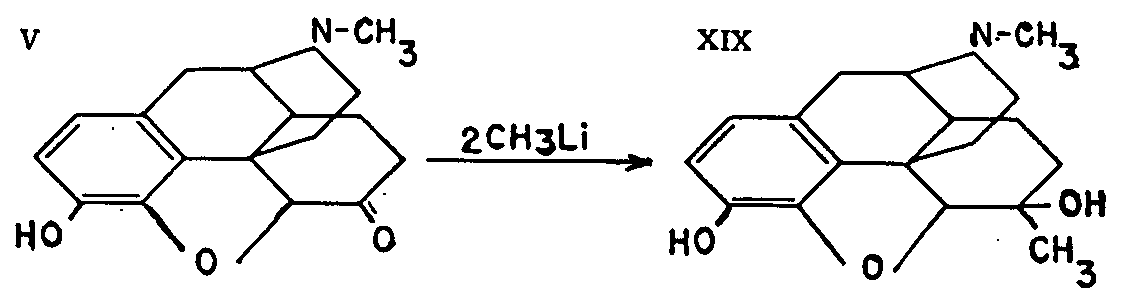
Small and Rapoport[6] are responsible for discovering this product (XIX). They demonstrated that the keto-groups of dihydrocodeinone and dihydromorphinone (V), which are virtually inert to Grignard reagents, react normally with methyl-lithium to produce the tertiary alcohols. 6-Methyldihydromorphine was obtained by the action of methyl-lithium upon dihydromorphinone. The most remarkable feature of this compound appears to be the duration of its analgesic action.
The preceding pages have been devoted to a description of the more important analgesics obtained by transformation of the natural alkaloids. It now remains to consider those analgesics which are independent of the natural bases and which have been made by total synthesis.
A large number of studies bear witness to the efforts which have been made to obtain pain-relieving drugs by complete synthesis. Although remarkable successes had been achieved, such as the discovery of synthetic local anesthetics and antalgics, no analgesic with an effect comparable to that of the opiates had been obtained by total synthesis until the last few years. "Only morphine and some of its derivatives can be regarded as true analgesics" wrote E. Fourneau in 1938, expressing the opinion of that day. It is true that the work, started in the United States in 1929 under the auspices of the National Research Council, directed by L. F. Small, included the synthesis of a certain number of true synthetic analgesics; but their analgesic effect, although real, had never approached that of morphine. A Public Health Service publication of 1941 summarizes the work done.[7]
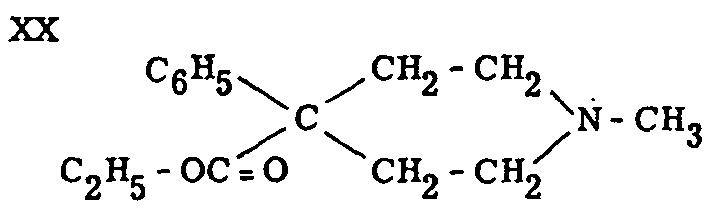
It was in 1939 that the situation suddenly changed, thanks to the sensational discovery of pethidine (dolantin, demerol) (XX) in Germany by Eisleb and Schaumann.[8] The synthesis of pethidine was not due to a priori considerations regarding the relations between constitution and activity, or to systematic research work on analgesics. It was achieved during work on antispasmodics, and the discovery of its analgesic effect was the result of a happy combination of luck and a pharmacologist's acumen.
This first completely synthetic analgesic demonstrated that analgesic properties were not necessarily dependent on a close chemical relationship with morphine, since pethidine is a comparatively simple phenyl-piperidine-carboxylic ester.
The experiments, which in number, quality and method are most representative of the tendency prevailing up to 1939, are those of the American school. Carried on simultaneously with a structural analysis of natural analgesics, they seem necessarily to bear the traces of their origin. They show a certain concern to duplicate part of the natural models, apparent in the special importance attached to certain structural elements or functional groups of morphine. Thus, amino-alcohols belong- ing to the phenanthrene, dibenzofuran and carbazole series were the favourite themes, varied by the introduction of a larger or smaller number of the functional groups present in the natural analgesics. The complexity of the morphine molecule stood in the way of a speedy and systematic investigation of the value of the various morphinic fragments, and the uncertainty which still persisted regarding its constitution was an additional obstacle.
It was in these circumstances that the chance discovery of pethidine brought about the revolution which released the imagination of chemists and opened new approaches to the problem. This revolution was marked by a remarkable simplification of the concepts hitherto accepted, the verification of which would have taken many years longer, and by the importance which was suddenly assumed by some parts of the morphine molecule considered less essential before. Schaumann realized only afterwards that the structure of pethidine could be related to that of morphine, if a special way of writing the two molecules was adopted. The formulations XXI and XXII are taken from Yonkman.[9]
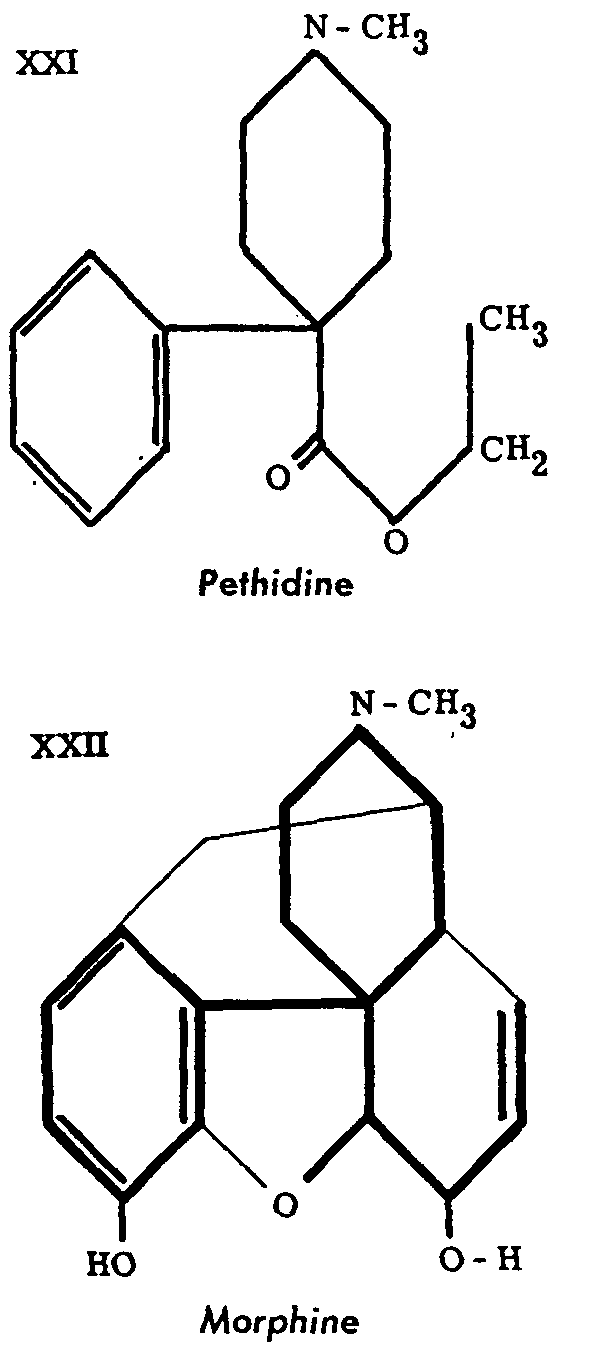
This relationship revived the interest of chemists in the study of synthetic analgesics, and thus opened up a whole unsuspected field of research. Its exploitation, which has been vigorously carried forward since the original discovery, has already produced such an ample harvest that only a brief summary of the work accomplished can be given. Excellent reviews like those of Bergel and Morrison,[10] some articles in the Barell Jubilee volume[11] or those of N. Eddy[12] and Wolff,[13] and the monograph of the New York Academy of Sciences[14] are available.
The work set in motion by the discovery of pethidine was directed at first quite naturally toward the exploitation of the initial success. Changes in the respective positions of the functional groups, modifications or displacement of subsituents, and the preparation of derivatives from the original molecule, were, and still are, important themes of research.
The articles referred to above give an account of the progress achieved in this direction and draw such conclusions as are possible regarding the new relations which appear between chemical composition and pharmacological activity. Those of Foster and Carman[15] and Eddy[12] are particularly instructive in this respect, and make a detailed discussion in this summary unnecessary.
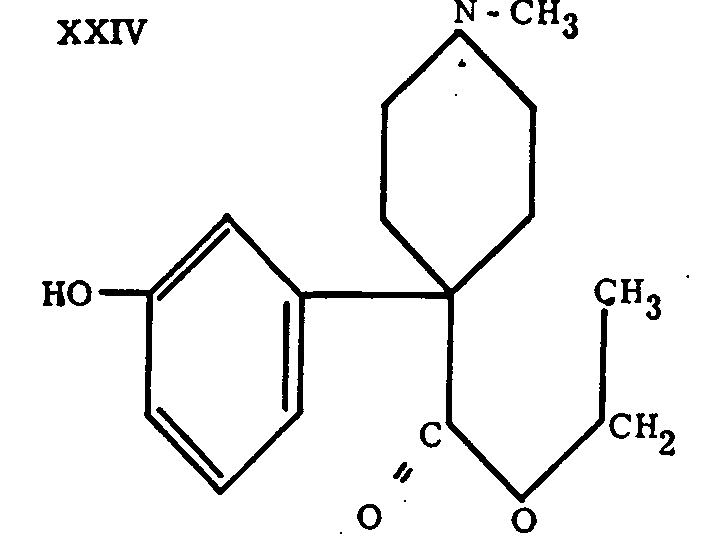
Among all the derivatives prepared in the pethidine series, we shall confine our attention in this general statement to "keto-bemidone" (XXIII), which results from a double modification of the pethidine molecule. The first consists of the introduction of a hydroxyl into the meta-position of the phenyl nucleus and produces bemidone (XXIV), the activity of which is comparable to that of the original molecule. The second modification replaces the carbethoxy group of bemidone by the ketonic grouping, -CO-C 2H 5, which increases the activity of the compound ten-fold.

The considerable activity of such a ketone obviously opened the way for a new series of completely synthetic analgesics, the chief of which is methadone (XXV). This compound, which, like keto-bemidone, was discovered by the I. G. research staff during the war, has the following formula.
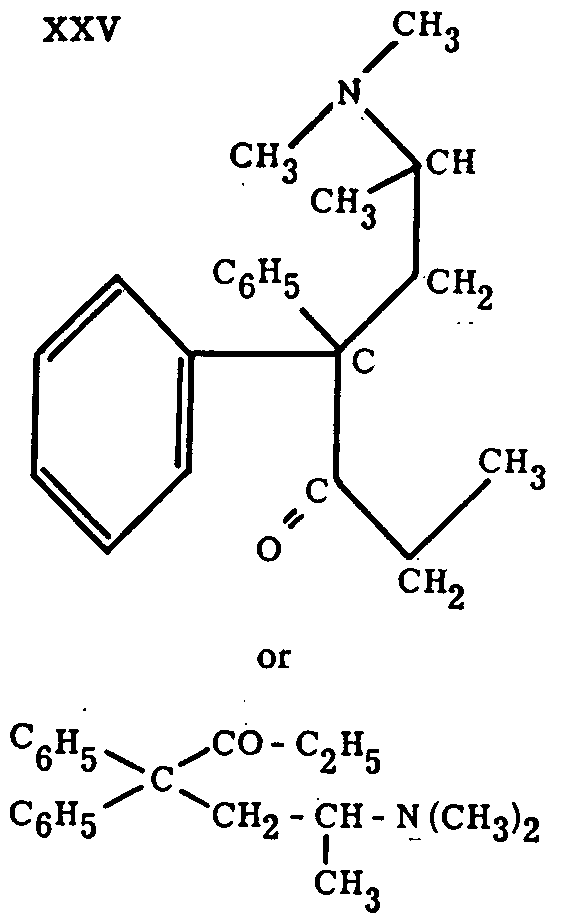
We recognize here the ketonic group -CO-C 2H 5 which is present in keto-bemidone and which makes this compound the bridge connecting the pethidine series with the methadone series. Nevertheless, methadone differs from keto-bemidone by the aliphatic character of its amino-nitrogen. The departure from the heterocyclic series is the result of the exploration by Bockmuehl and Ehrhart[16] of a series of compounds containing the group below, instead of the piperidine nucleus; some of those compounds show both anti-spasmodic and analgesic effects. Methadone is therefore reminiscent of two previous discoveries, one demonstrating the importance of the ketonic function, which is foreign to natural anal- gesics, and the other the possibility of replacing the piperidine nucleus by an aliphatic chain carrying a substituted amino group.
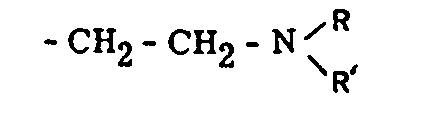
The analogy of methadone with morphine is even more remote than that of pethidine. Yet its formula, given above, brings out certain points of resemblance. These are confirmed, according to Bergel and Morrison, (loc. cit.) by a comparison of the molecular models, which shows the spatial similarity of methadone to morphine and the phenyl-piperidines.
As in the case of pethidine, methadone was the starting point for a vast amount of research on related compounds, which are reviewed in the article by Eddy (loc. cit.; especially Table III, page 249).
Before leaving the methadone series in these general remarks, let us note that no noticeable increase in its activity has been obtained by the modifications made in its molecule, except by optical resolution.
The two series which we have passed in rapid review were only attached to the morphine series by a postieri considerations, and do not even derive from it historically. This is not true of the morphinane series, which we shall now consider. The very name of the first product (XXVI), clearly emphasizes its relationship with the natural alkaloids. This is no longer a fragment of the morphine structure, but rather its complete skeleton as the following formula shows:
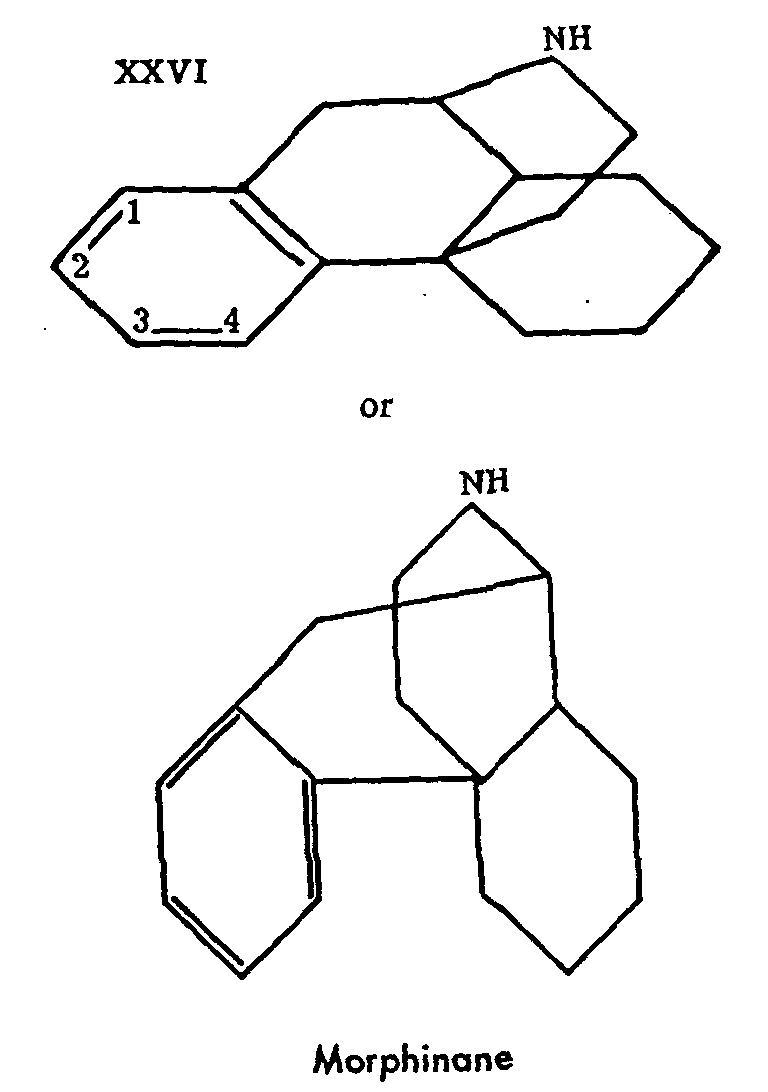
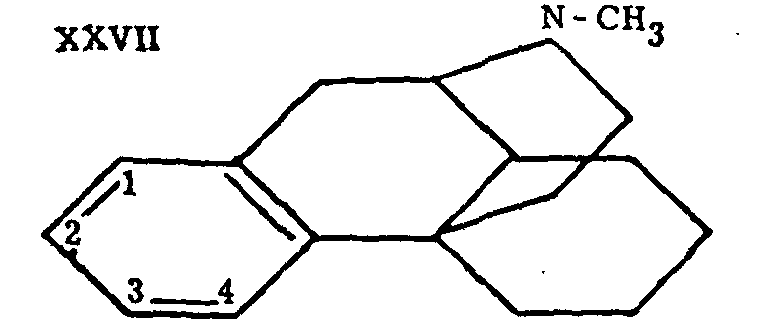
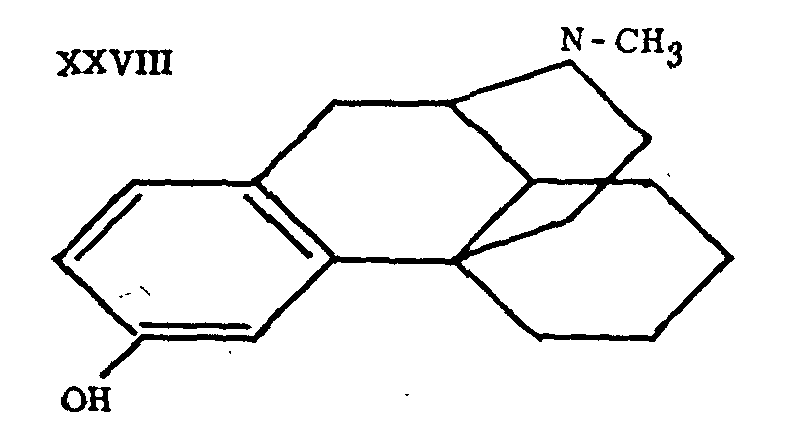
The brilliant successes so rapidly achieved in this series are due chiefly to Grewe[17] ; the synthesis of morphinane marks the first memorable stage of this work. Other authors had attacked the problem of the synthesis of compounds containing the complete morphine skeleton in the hope of preparing the way for the synthesis of morphine. The review of Bergel and Morrison gives an account of these experiments, prior to those of Grewe. Their interest is now mainly historical. The work of Grewe and his associates or competitors will be discussed later in a special chapter. Up to now, its culminating point has been the synthesis of the l-isomer of tetrahydrodesoxycodeine (II), which constitutes the first example of the synthesis of a compound identical with a derivative of a natural analgesic still possessing the complete skeleton of morphine. Grewe's success is not only outstanding in the chemical field, it far transcends it, by throwing new light on the relations between the structure and the activity of analgesics. Thus N-methyl-morphinane (XXVII), without possessing the oxygen bridge, the hydroxyls or the double bond of morphine, has proven, contrary to all expectations, to have an undeniable analgesic effect, which is strengthened and prolonged by the simple introduction into position 3 of a hydroxyl, analogous to the phenolic group of morphine, to such an extent that 3-hydroxy-N-methyl-morphinane (XXVIII) becomes a rival to morphine.
It would appear that a comparative examination of the natural, and the partially or completely synthetic analgesics ought to show clearly to what structural or functional characteristics the analgesic power and its properties should be attributed. These lessons, however, have been neither as numerous nor as valuable as might have been hoped. The stereochemical aspect of the problem of the relationship between the chemical structure and the analgesic activity has been discussed in the review of Bergel and Morrison. To enter into it here would exceed the limits of this article without adding anything to the excellent account of the English authors. We shall therefore content ourselves with reproducing the conclusions of N. B. Eddy at the end of his 1950 paper which sums up our present knowledge:
The presence of a tertiary amino-nitrogen seems to be essential in an analgesic compound. This nitrogen may be part of a heterocycle (piperidine, tetrahydroquinoline, morpholine, pyrrole...) or of an open chain.
The presence of a quaternary carbon linked through -C-C- with the amino-nitrogen has been noted hitherto in analgesics in the morphine, pethidine and methadone series. Its presence, and the relationship to nitrogen described above, might not be absolutely necessary, but at least they seem to be responsible for the maximum intensity of the analgesic effect.
Certain substitutions of functional groups for others, listed in Eddy's article, produce changes of the same type and of the same extent if carried out in the different analgesic series.
The toxicity and the analgesic effect are not necessarily affected to the same extent or in the same way by the various modifications described.
Unfortunately, no analgesic has been obtained which does not induce habituation and addiction.
The three series of completely synthetic analgesics considered so far, the pethidine, methadone and morphinane series, do not include all the analgesics discovered in the course of recent research. A number of these are not covered by any systematic classification. We shall therefore have to consider them in a separate group.
Leaving now this chapter of general considerations, intended to facilitate the understanding of the following chapters which are devoted to the chemistry of the various series of completely synthetic analgesics, we shall abandon the chronological order to adopt one which is more logical from the chemical point of view. We shall begin with the morphinane series, which has the same fundamental structure as the partially synthetic analgesics. There will thus be a more natural transition to the pethidine and methadone series, the relationship of which to the natural analgesics is becoming increasingly remote. The synthetic compounds which do not fall easily into any classification will be examined at the end.
The importance attributed to the various series will not be the same. It is our belief that it is of little interest to paraphrase or recapitulate the excellent articles which have already appeared and to which we have referred above. The morphinane series is the only one which apparently has not yet been the subject of a review, and will therefore be discussed in greater detail than the rest.
The first group of synthetic substances to be discussed consists of those which possess the same carbon-nitrogen skeleton as morphine itself. The synthesis of such structures was first achieved by Grewe in 1946.[17] Before describing this work, however, it may be interesting to mention a few forerunners.
A number of authors have synthesized compounds which duplicate only certain characteristic parts of the morphine skeleton rather than the entire structure.
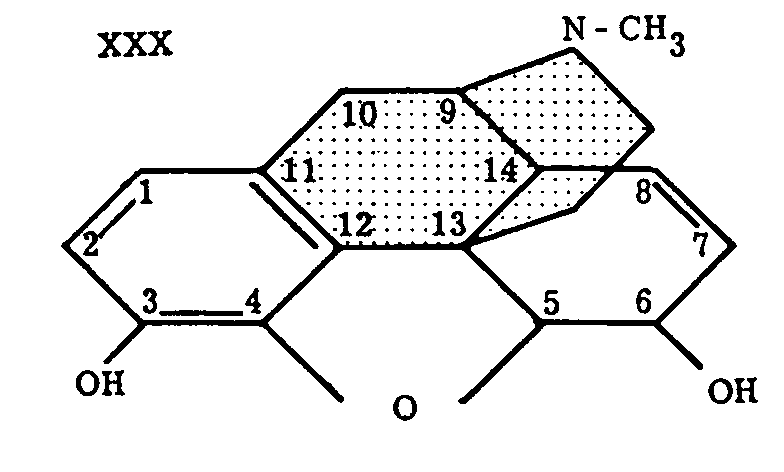
Since the ring system, 2-azabicyclo-[3,3,1]-nonane (XXIX, R=H), is present in the formula of morphine (XXX), the name "morphan" was proposed for the above structure by Barltrop,[18] who describes the synthesis of some of its derivatives, such as XXXI and XXXII.
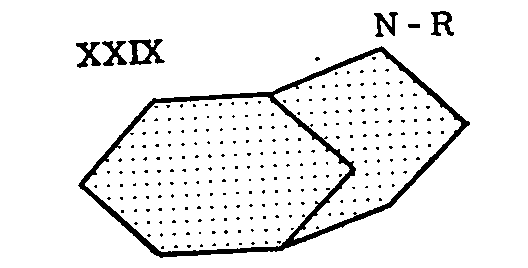
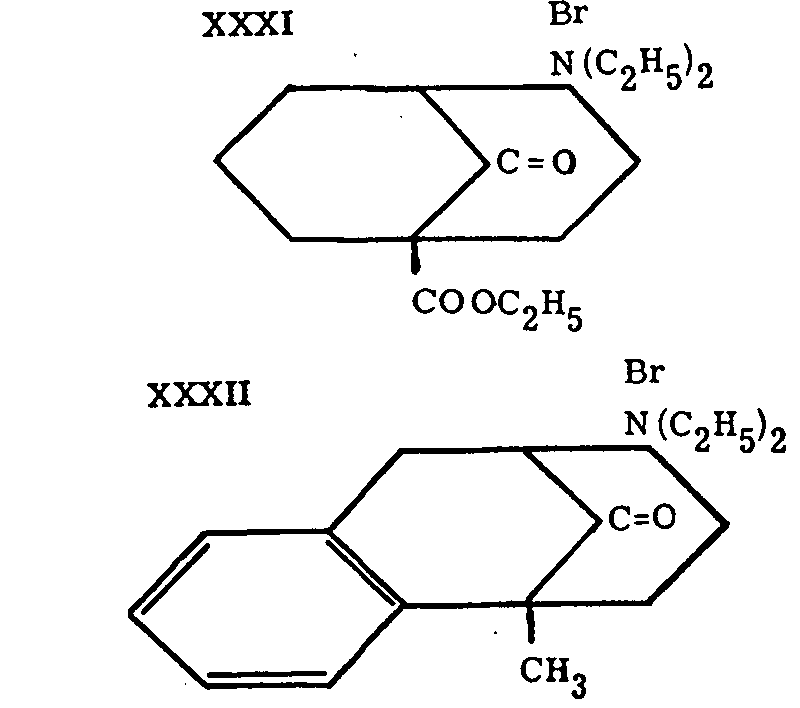
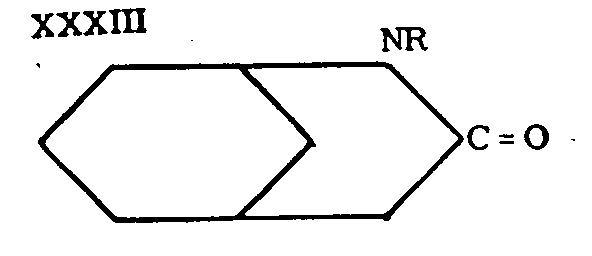
Subsequently, Cronyn[19] and Ginsburg[20] obtained morphan itself and its N-ethyl derivative (XXlX, R= C 2H 5) through reduction of the corresponding lactams (XXXIII). The lactams had a certain analgesic activity, while morphan itself and its N-ethyl derivative were inactive.
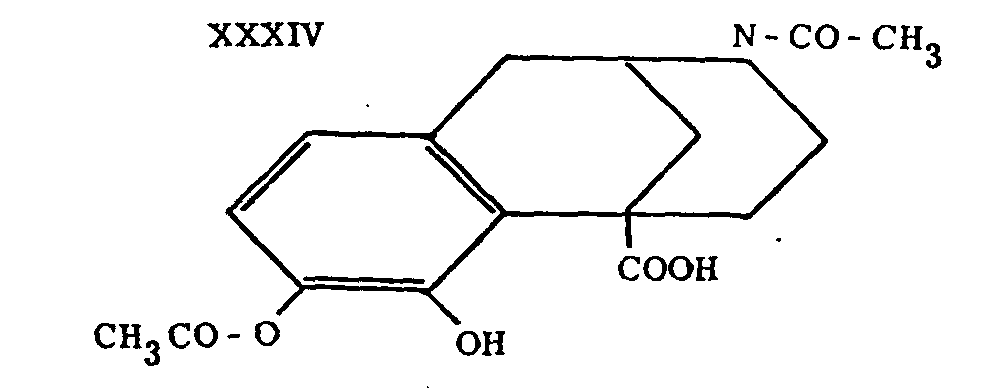
Related compounds such as XXXIV were prepared by Horning and Schock.21
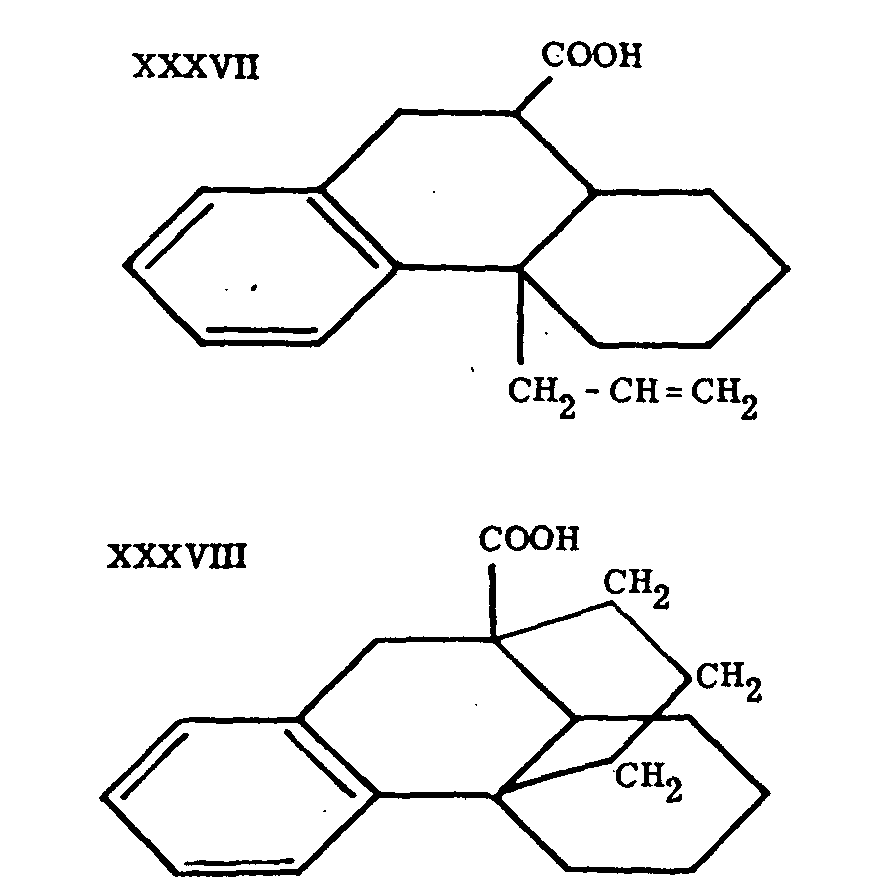
Attempts in another direction aimed at synthesis of the partially hydrogenated phenanthrene system with a substituent in position 13.[22] Similar compounds were prepared by Grewe[23] at earlier stages of the work that later culminated in the synthesis of substances containing the entire C-N skeleton of morphine. Compounds such as XXXV and XXXVI were obtained, but at tempts to prepare XXXVII led to XXXVIII instead. This cyclization to a substance with a ring system similar to that of morphine itself induced Grewe to investigate the cyclization of heterocyclic systems analogous to XXXVII.
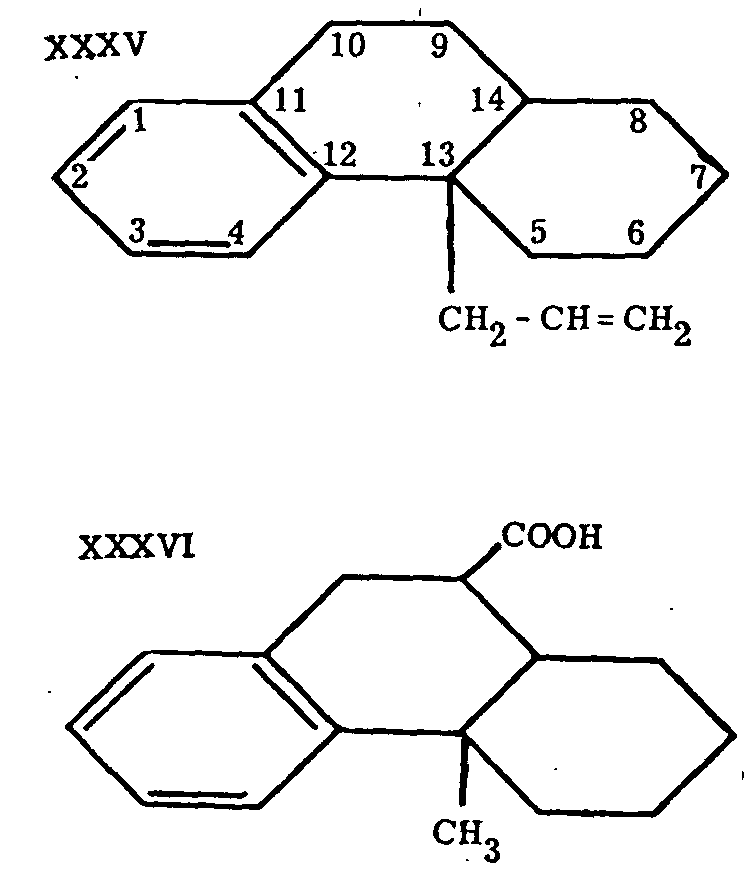
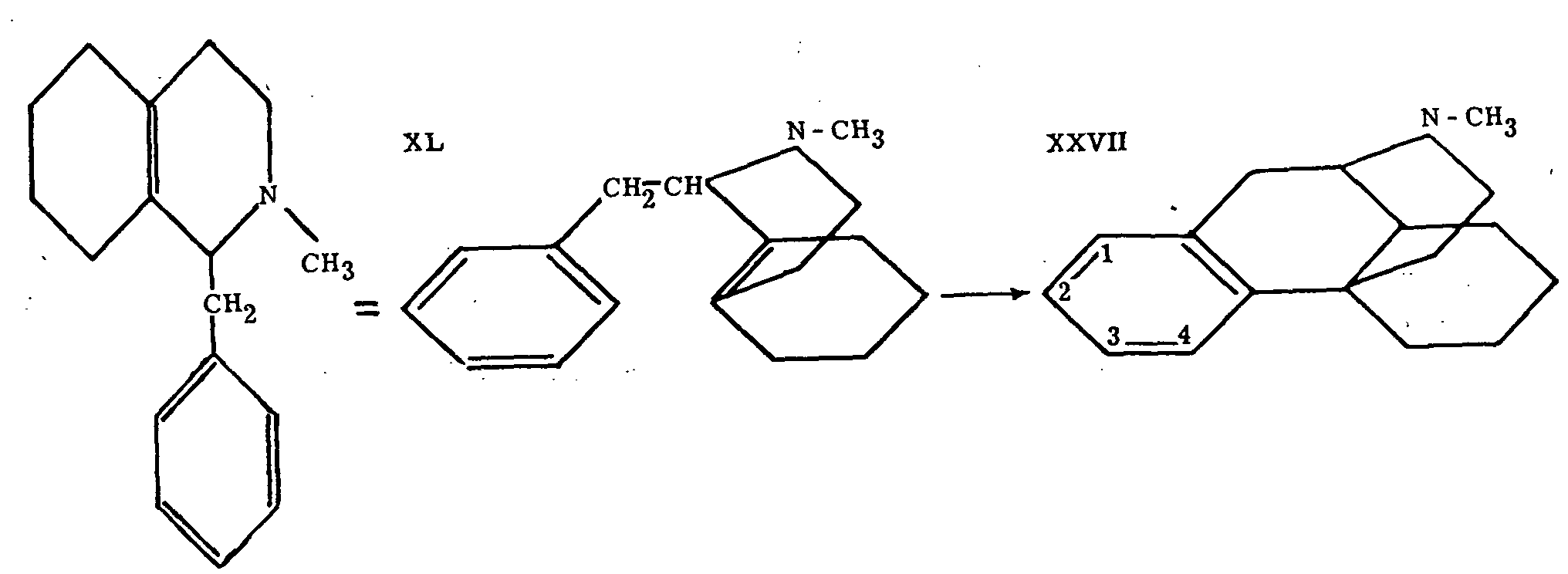
A comparatively simple synthesis of 5, 6, 7, 8-tetrahydro-isoquinoline (XXXIX) was worked out (see Chart I); this compound was further transformed to the desired N-methyl-l-benzyl-octahydro-isoquinoline (XL) in the manner shown in Chart I, if Ar=C 6H 5 The formula of this last compound can be rewritten in such a way that its relationship to the morphine type of structure becomes readily apparent. Although the required cyclization to XXVII, N-methylmorphinane, seems without a close analogy, it proceeded smoothly on heating XL with concentrated phosphoric acid to 150o, yields better than 50 per cent of XXVII being obtained, together with smaller quantities of two byproducts, one of which is identical with a compound recently synthesized by an entirely different route.
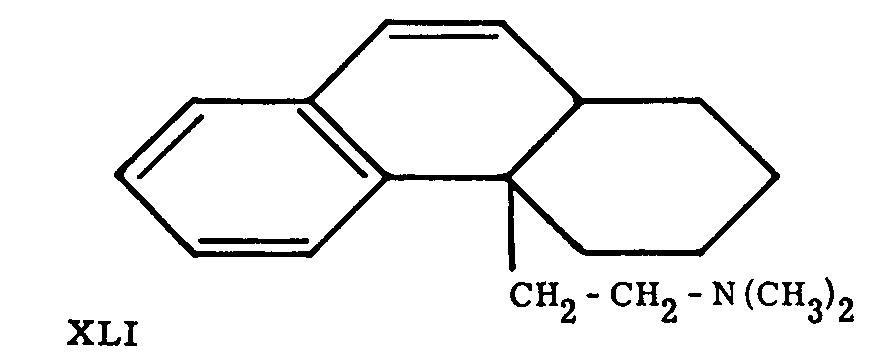
Grewe proposed the name "N-methyl-morphinan for the main product (XXVII) of this brilliant synthesis; its structure was proved by exhaustive methylation to the des-base XLI, which was readily converted to phenanthrene by heating with Pd-charcoal, and reduced catalytically to a dihydro derivative identical with a compound prepared by an independent synthesis.
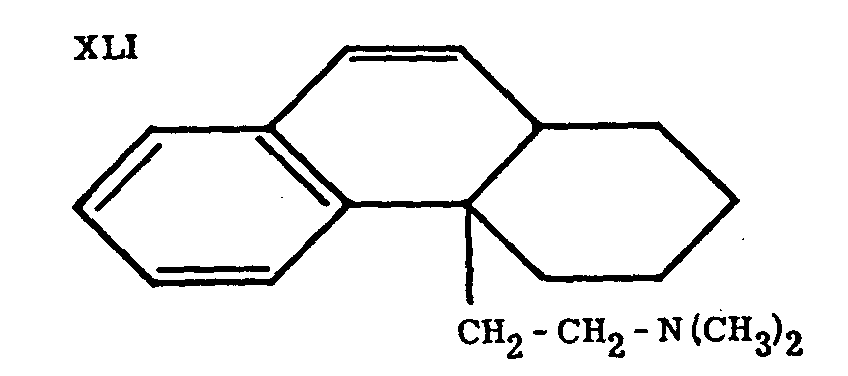
Action of BrCN on N-methylmorphinane yielded the parent compound of the whole series, morphinane (XXVI).
On pharmacological investigation, N-methylmorphinane showed pronounced analgesic activity, a very surprising property for a compound of morphine-like structure lacking the oxygen bridge.
This analgesic activity is much more pronounced in 3-hydroxy-N-methylmorphinane (XXVIII), which was obtained by Grewe, Mondon and Nolte[2] and by Schnider and Griissner[24] through cyclization of the compound XLII and demethylation (Grewe et al. achieved both reactions in one step by heating with concentrated HBr). The Swiss authors[24] also prepared 3-hydroxy-N-methylmorphinane by nitration of N-methylmorphinane and subsequent reduction and diazotization. The high, rather long-lasting analgesic activity of this compound (also referred to as morphinan, dromoran and NU 2206) has led to its thorough pharmacological and clinical investigation.
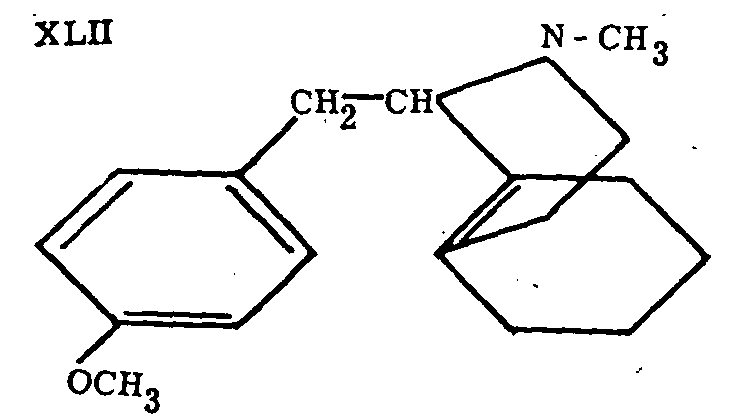
Schnider and Grussner (loc. cit.) also prepared 2-(or 4-) hydroxy-N-methylmorphinane (XLIII or XLIV; it was not established which isomer is formed), but found it entirely inactive. The same is true of compounds of the morphinane series having groups other than methyl on the nitrogen.
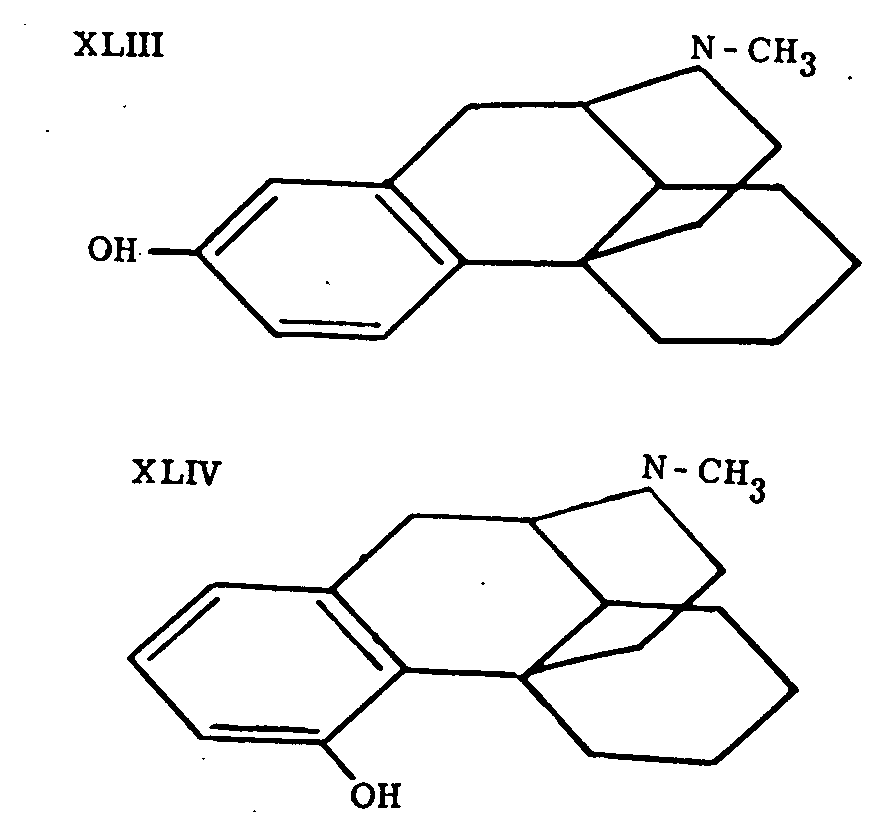
Further important progress in this field is due to Grewe, Mondon and Nolte (loc. cit.), who succeeded in synthesizing the compound XLV and who identified its levo-isomer with tetrahydro-desoxycodeine (II) prepared from codeine.* This constitutes the first total synthesis of any compound of the morphine series which is also accessible through transformation of the natural alkaloids by methods which presumably do not alter the fundamental ring system. The identity of the synthetic substance with the one derived from codeine provides very strong support for Gulland and Robinson's morphine formula (I).
For the synthesis of compound XLV, the route utilized in the synthesis of N-methylmorphinane could not be used, since the required compound Ar-CH 2-MgHal with Ar=3, 4-(CH 3O) 2 C 6H 3- is not accessible; the authors therefore attempted first to start with N-methylmorphinane (XXVII) itself, trying to introduce a halogen atom into position 4, which was to be exchanged subsequently for an OH group. However, it was found that halogen invariably enters position 2 first, 2, 3-dihydroxy-N-methylmorphinane being finally obtained.
* United Nations Bulletin on Narcotics, vol. II, No. 2, page 19, and page 12, this issue.
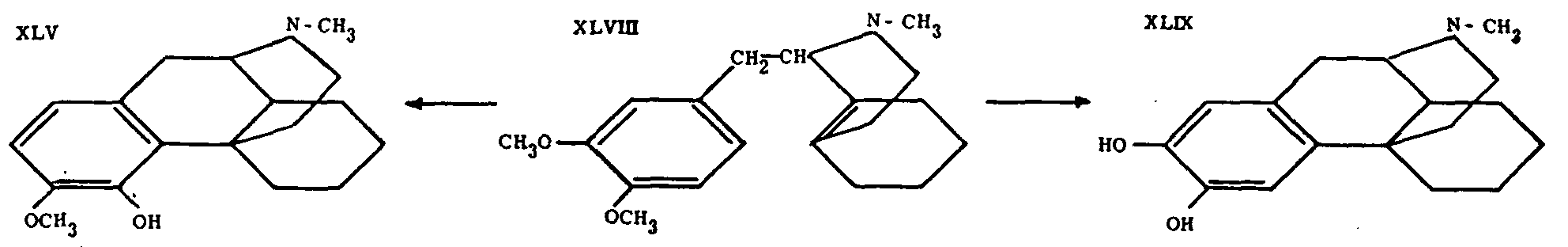
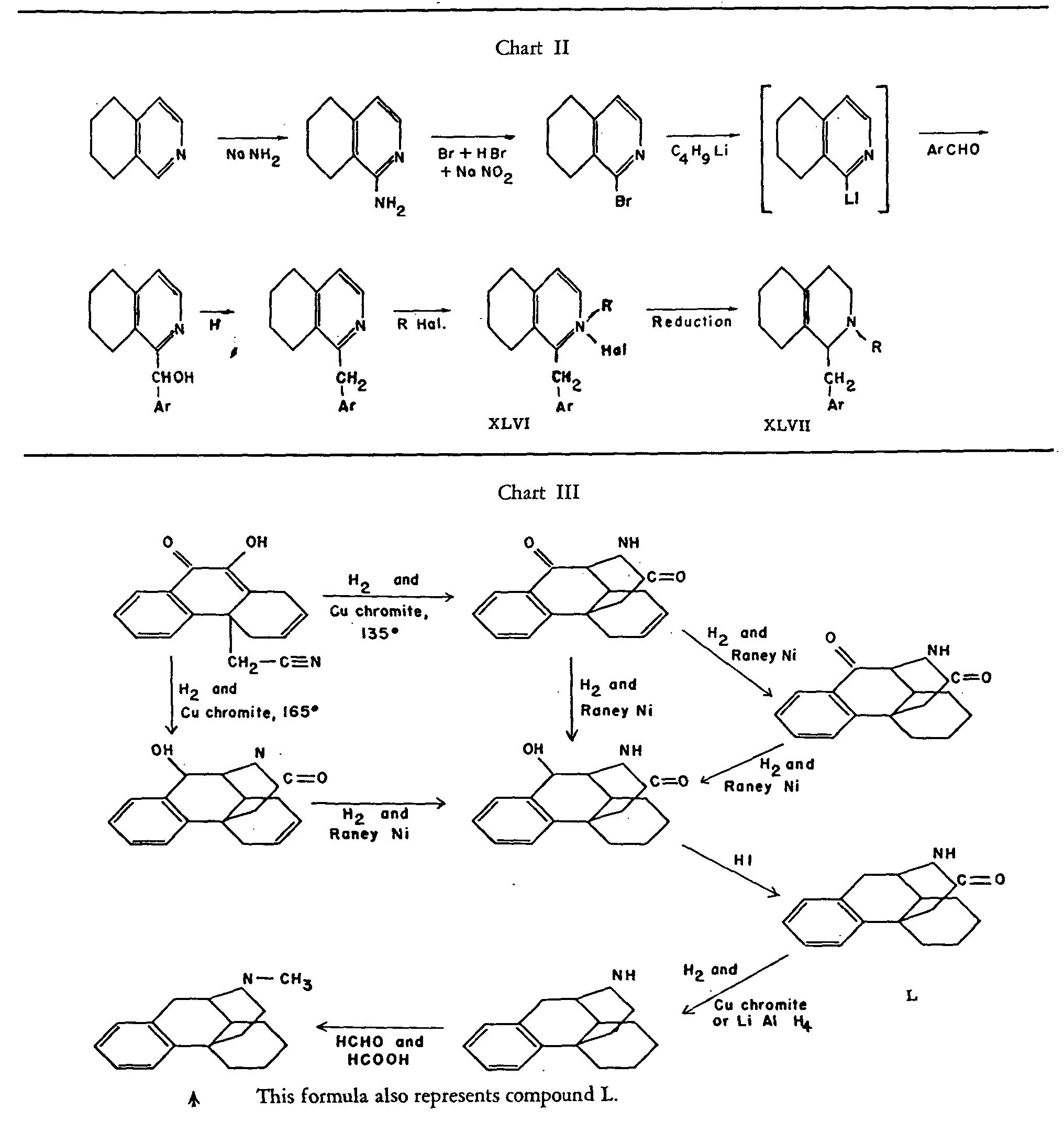
A new sequence of reactions had therefore to be worked out (See Chart II). Following this general method, morphinane and 3-hydroxy-N-methylmorphinane were first prepared. In the first case, the intermediate chlorobenzylate of 1-benzyl-tetrahydro-isoquinoline (XLVI, R=Ar=C 6H 5) was reduced catalytically to 1-benzyl-octahydro-isoquinoline (XLVII, R=H, Ar=C 6H 5), which was then cyclized to morphinane. Finally, the synthesis with R=CH 3 and Ar=3, 4-(CH 3O) 2C 6H 3- yielded XLVIII. This, on heating to 130° with concentrated HCl, underwent demethylation in position 4 and cyclization simultaneously to give the desired 4-hydroxy-3-methoxy-N-methylmorphinane (XLV).
This product was shown to be identical with the racemate obtained by mixing the I- and d-forms of tetrahydrodesoxycodeine (II), prepared by a series of transformations starting with natural codeine and sinomenine, respectively. Furthermore, resolution of the synthetic racemate XLV via the tartrate yielded the 1-isomer, identical with l-tetrahydrodesoxycodeine prepared from codeine.
It is remarkable that in the cyclization of N-methyldimethoxybenzyloctahydro-isoquinoline the alternative ring closure to the 2, 3-substituted morphinane does not seem to take place; it does so, however-at least in part-if more strenuous conditions are used for the reaction. Because of its sensitivity, the resulting base XLIX was not isolated as such, but as its dimethylether.
It is equally remarkable that among the possible diastereomers of XLV only the one corresponding in its configuration to the natural alkaloids has been obtained. However, as has already been mentioned on page 20, this does not apply to the synthesis of N-methylmorphinane, where two isomers of the main product were produced together with the latter. That one of them is actually a stereoisomer of N-methylmorphinane is shown by the recent work of Gates and co-workers.[25]
These authors carried out the sequence of reactions outlined in chart III; they found that the final product (L), an isomer of N-methylmorphinane, has the same constitution as this compound and is identical with one of the two by-products in Grewe's synthesis
The structure of the base synthesized by Gates et al., for which the name N-methyl-isomorphinane was proposed, has been established by its exhaustive methylation to the des-base, a stereoisomer of XLI ( see p. 23), which was converted to 1,2,3,4-tetrahydiophenanthrene on distillation with zinc dust.
It is stated by Gates et al. that N-methyl-isomorphinane shows considerable analgesic activity in animal tests.
Since the completion of the manuscript of this article, several papers in the field of morphinane and its derivatives have appeared, which should be mentioned briefly.
A short review of this subject by A. Mondon has become available. [26]
Two United States patents[27] cover the synthesis of N-methylmorphinane and describe many derivatives and related compounds.
Schnider and Hellerbach[28] give a new synthesis of 1-benzyl-octahydroisoquinolines (XL) which does not require the use of benzyl-magnesium halides. This route is therefore well adapted to the preparation of such compounds of type XL, which could not be made by the original procedure of Grewe because of the impossibility of preparing the required Grignard reagents. By cyclization of their benzyl-octahydroisoquinolines, Schnider and Hellerbach obtained several new derivatives of morphinane, including d,l-tetrahydrodesoxycodeine.
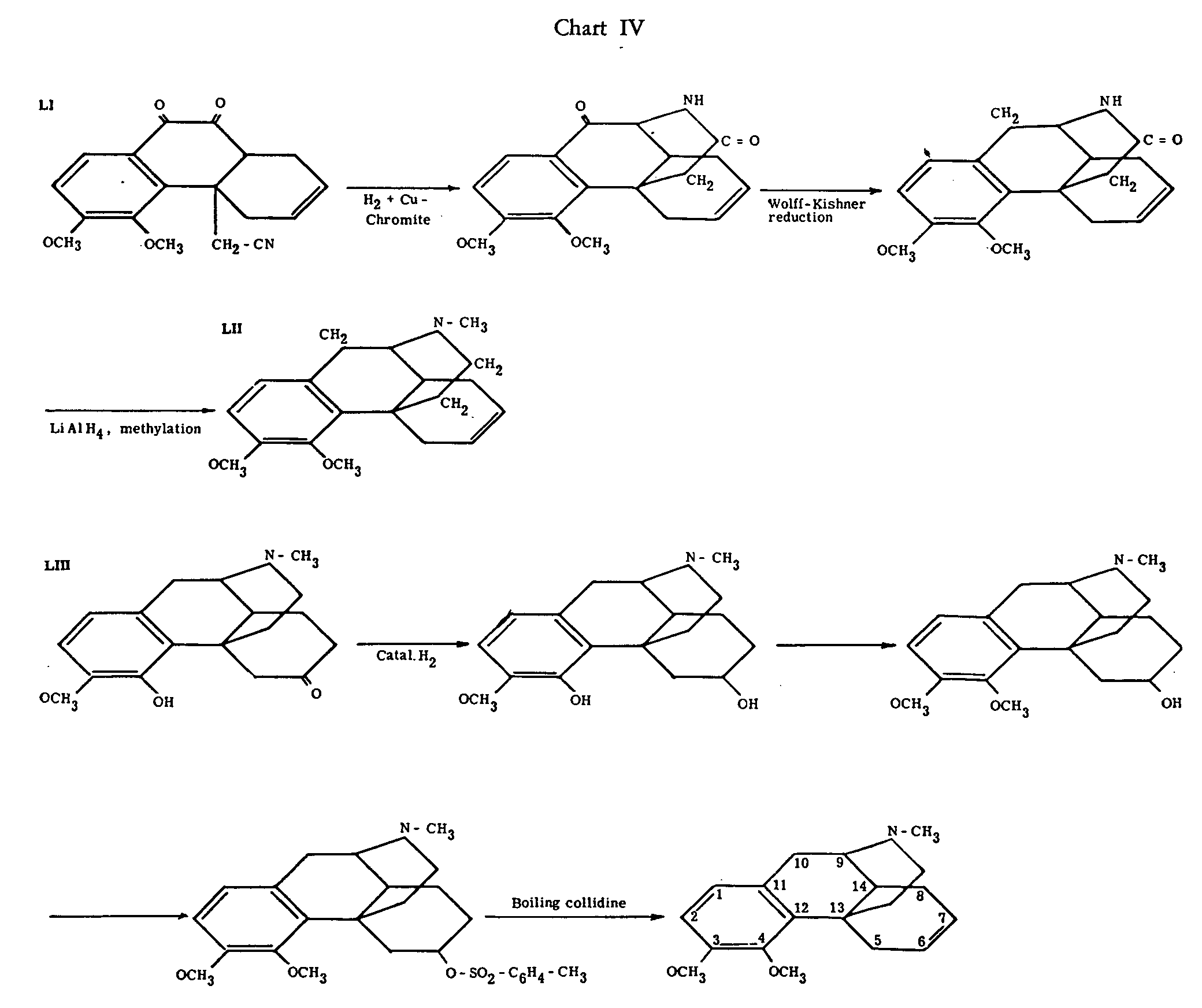
Gates and Tschudi[29] made important progress in the isomorphinane series. They succeeded in cyclizing LI in a way analogous to that formulated in chart IV for the corresponding methoxyl-free substance, and in converting the reaction product in several steps into LII. This racemic compound was compared with material prepared by the sequence of reactions outlined in chart IV, starting with either dihydrothebainone (LIII) or B-dihydrothebainone. This latter ketone had been prepared by Small and Browning[30] and formulated as the epimer of LIII with opposite configuration at C-14. Gates and Tschudi compared the optically active products derived from dihydrothebainone and β-dihydrothebainone respectively with the synthetic racemate by infra-red spectroscopy, and found the spectrum of LII to be identical with that of the compound from β-dihydrothebainone.
This identity provides excellent support for the correctness of the structure of N-methyl-isomorphinane, and its interpretation as the epimer of N-methyl-morphinane. Beyond that, the fact that the product of LII of a cyclization of the C-13-substituted phenanthrene LI has been found identical with a derivative of natural codeine, shows that in the latter the nitrogen-containing chain must equally be attached to C-13. The unequivocal establishment of this point by degradative methods had constituted the main difficulty in the research on the structure of codeine.
The history of the synthesis of pethidine, and of the discovery of its analgesic properties by Eisleb and Schaumann[8] has been related above.
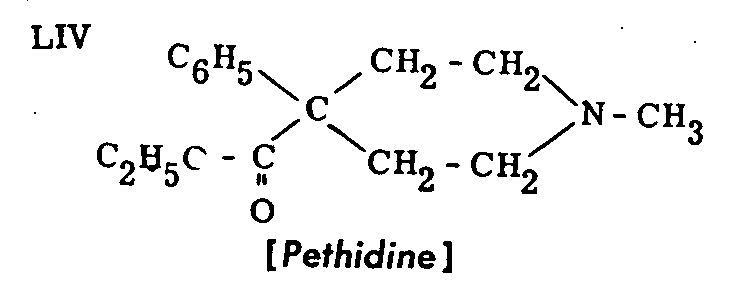
Pethidine was originally named "dolantin" and is also known under the synonyms demerol, dolantal, meperidine, isonipecaine, etc. It is the ethyl ester of N-methyl-4-phenyl-piperidine-4-carboxylic acid. Its chemical formula can be written as LIV, or LV (in Chart V).
Pethidine was first synthesized by the condensation of benzyl cyanide with N-methyl- β, β'-dichloro-diethylamine in the presence of sodamide, saponification of the nitrile group and esterification of the carboxylic acid thus obtained. See chart V.
Several other methods of synthesis have been suggested since then; their principal aim was to avoid the use of the powerful vesicant, N-methyl-dichloro-di- methylamine. The German method which was made public after the war, and which was described in American and British technical reports[31] on captured documents, uses the less-dangerous N-benzyl- β, β'-dichlorodiethylamine. The substitution of methyl for benzyl then takes place during a final reaction which utilizes reduction by hydrogen in the presence of palladium and formaldehyde. Chart VI illustrates that method, which has an overall yield of about 25 per cent.
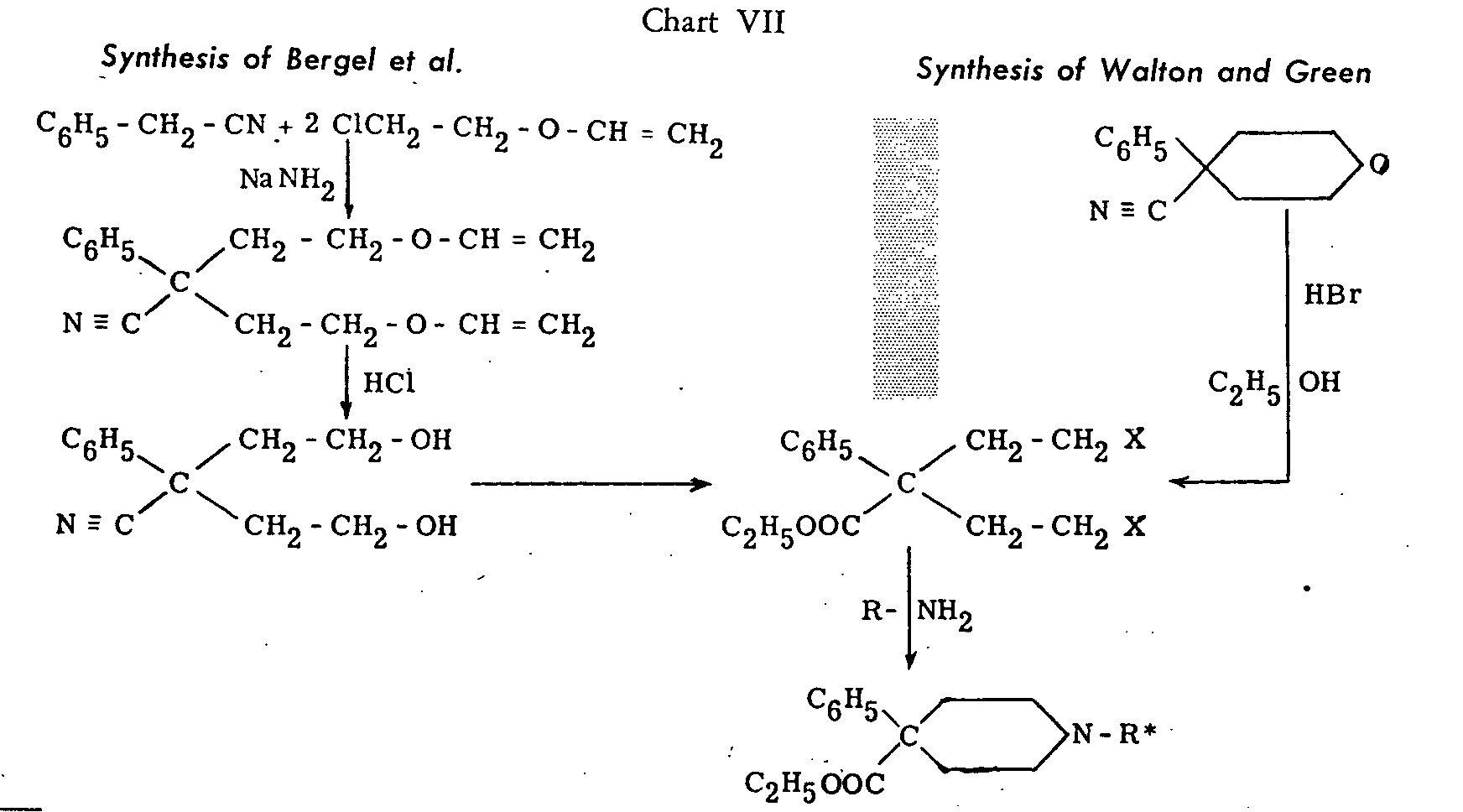
Two other methods successfully used by Bergel and his collaborators[32] and by Walton and Green[33] are illustrated in chart VII ( see p. 26).
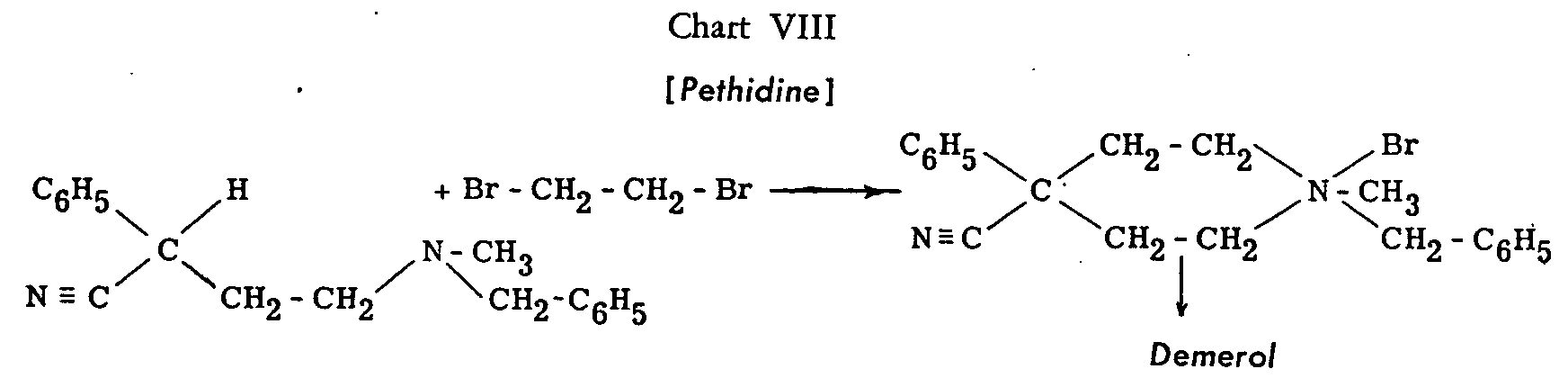
Of the other proposed methods of synthesis, we shall only mention one for which Ciba holds a British patent[34] and in which pethidine is obtained through the hydrogenolysis of a quaternary ammonium salt (Chart VIII) ( see p. 26).
Recently, radioactive pethidine, labeled in the N-methyl group, has been prepared. This compound was obtained through the methylation of 4-phenyl-4-carbethoxypiperidine by means of a mixture of radioactive formaldehyde and formic acid.[35]
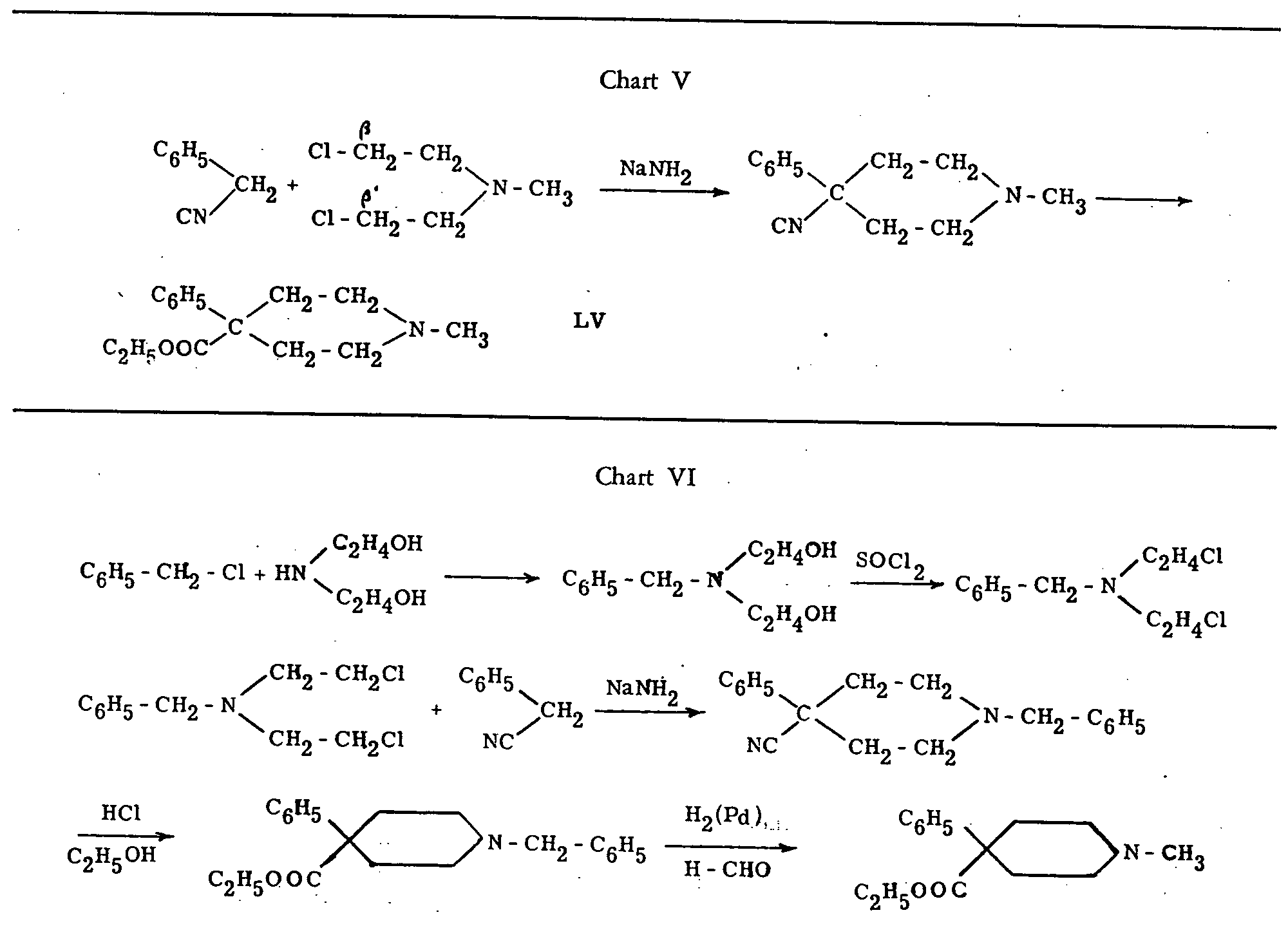
* R is CH 3 in the case of pethidine.
The physical and Chemical properties of pethidine are not of sufficiently general interest to be described here The pharmacology of pethidine has been described by Yonkman in an article published in 1948.[9] Although pethidine is a less powerful analgesic than morphine, it has the advantage of not depressing the respiratory center; it is therefore valuable in obstetrics. The initial belief that pethidine is not habit-forming has not been confirmed by subsequent observations.
The discovery of pethidine was rapidly exploited. As early as 1940 Schaumann himself[36] was able to report on pharmacological tests dealing with over forty compounds synthesized by Eisleb.[37] Various other research workers displayed such activity that by 1948 the pethidine series comprised over 200 members. To give even a condensed account of this work is unnecessary since the above-mentioned reviews by Bergel and Morrison or Eddy are available. To conclude this discussion of the pethidine series, it will be therefore sufficient to classify not the derivatives themselves but the various modifications undergone by the original molecule, according to the molecular or functional group they affect. Evidently, some of the transformations were brought about separately, while others were combined with each other.
We shall examine three groups of modifications of pethidine according to whether they affect the benzene ring, the piperidine ring or the ester function, each of them comprising a few sub-divisions.
Various subsituents were introduced into different positions; for instance, one methyl in the ortho position; a hydroxyl, free, etherified or esterified, in the meta or para position. Bemidone (XXIV) is among the derivatives of this group, the synthesis of which sets out from m-methoxybenzyl cyanide according to the method used in the preparation of pethidine; the only difference is that hydrolysis of the nitrile group and demethylation are achieved simultaneously by the action of HBr.
The phenyl group was separated from the quaternary piperidinic carbon by insertion of a carbon chain.
The benzene nucleus was replaced by either hydrogen or various aliphatic, aromatic or alicyclic radicals. In most cases, these transformations were made simultaneously with the replacement of the carboxyester group by esters derived from a piperidinic carbinol in position 4. In the latter case, the synthesis sets out from the corresponding 4-piperidone, utilizing LiAlH 6, or organo-magnesium or -lithium compounds.
The methyl attached to the nitrogen was replaced by other radicals. Its replacement by butyl, accompanied by the change from the carboxylic series to the carbinolic series, with esterification by propionic acid, increases the analgesic activity.
The respective positions of piperidine nitrogen and quaternary carbon were modified. The series with the quaternary carbon in the 3 rather than in the 4 position is generally known as the iso or βseries. The carbon having become asymmetric, the synthetic racemate may now be resolved into two enantiomorphs, the more active of which is the levorotatory isomer.
The addition of substituents to the piperidine ring made it possible to show that the activity is increased by the presence of a methyl in position 3.
The ethyl radical was replaced by numerous other radicals. Such changes usually produced a considerable decrease in analgesic activity, except in a few rare cases (isopropyl, allyl, n-propyl).
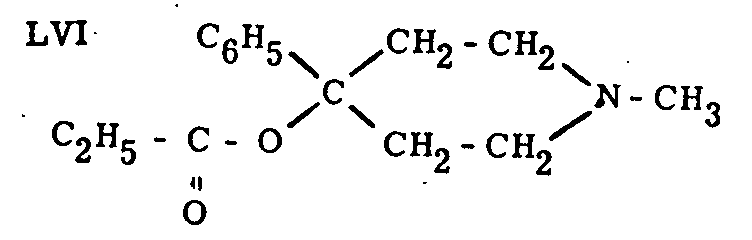
The carboxylic group was replaced by a hydroxyl. The carbinol thus obtained was acylated by various acids. Propionic acid led to the most active compound of the series (LVI), about five times more active than pethidine itself.
The oxethyl part of the ethyl carboxylate group was replaced by various radicals, thus giving rise to the ketonic series; its most important representative, ketobemidone (XXIII) ( ?. 17) has already been mentioned. Replacement by -NH 2 was also tried; the resulting amides were active.
The carboxyl group was shifted from its position 4 on the piperidine ring, with loss in activity.
The carboxyl group was replaced by hydrogen or certain radicals such as phenyl, or even by a sulfone group.
Certain transformations of the pethidine molecule affect several of its functional groups; some examples, such as keto-bemidone, have already been mentioned in this paper.
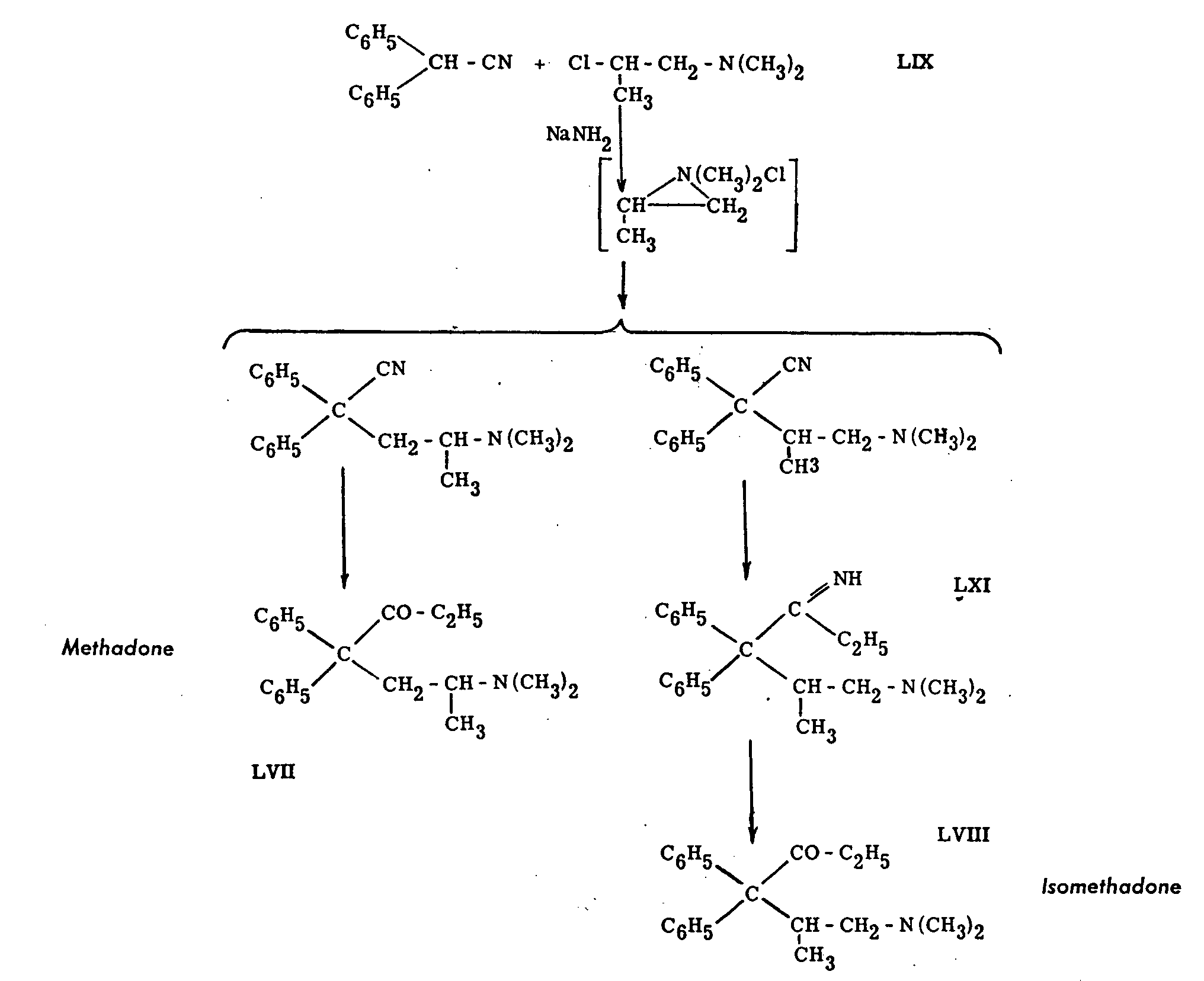
Methadone (LVII, in Chart IX) resulted from research in the laboratories of I. G. Farbenindustrie A.G. at Hoechst-am-Main, Germany. It was referred to as compound 10820, or amidone; its value, apparently not quite appreciated by its discoverers, became known through reports of Allied investigators.[38] The powerful analgesic action of the compound has led to great interest in its pharmacology; it has been introduced into medical use in the United States under the name "methadone'', in England as "amidone." Other names used for this product are: dolophine, adanone, physeptone, polamidone, AN-149, etc.
D,1-methadone is stated to be a stronger analgesic, weight by weight, than morphine, and to resemble it in most respects, unfortunately also in addiction liability; certain pharmacological differences, however, have been found. From the very extensive literature on the pharmacology of methadone, only a few leading references can be given here: Chen, Annals of the New York Academy of Sciences, 51, 83 (1948); Eddy, Touchberry and Lieberman, Journal of Pharmacology and Experimental Therapeutics, 98, 121 (1950); Denton and Beecher, Journal of the American Medical Association, 141, 1146 (1949).
The synthesis of methadone proceeds by the steps shown in Chart IX, leading finally to the two end products, methadone and isomethadone (LVIII).
The chemistry of the reactions involved has been the object of much study; the rearrangement occurring in the synthesis of methadone is generally explained by assumption of reaction of the chloro-amine (LIX) in the cyclic form indicated in the flow sheet, The constitution of the various compounds has been demonstrated by degradative reactions and partly by independent syntheses.[39]
The resolution of the racemate, methadone, into the antipodes has apparently been studied in Germany. It has subsequently been achieved by several groups of workers: Thorp, Walton and Ofner,[40] Brode and Hill,[41] Larsen, Tullar, Elpem and Buck,[42] Howe and Sletzinger.[43] Comparison of the activity of the antipodes by several authors has shown that the l-isomers of methadone and isomethadone are much more potent analgesics than the d-isomers.[44]
As was the case with pethidine, the success of methadone led to intense study of many chemically related compounds; only a few of the more important results can be mentioned here. The variations of the basic structure can be logically divided into the following groups:
Modifications of the aromatic rings.
Modifications of the nitrogen-containing side chain.
Modifications of the ketonic side-chain.
For more extensive discussions of the changes made, and the regularities found, see the articles by Chen and Eddy (loc. cit.).
1. Modifications of the aromatic rings
The introduction of substituents into the aromatic rings, or their replacement by other cyclic structures, led to diminution or disappearance of analgesic activity. Already the workers at Hoechst made the unexpected observation that introduction of one meta-OH group into a compound of the methadone series yielded a product having only a fraction of the activity of the parent substance; this is in striking contrast to the high activity resulting from the analogous change in the molecule of pethidine to give keto bemidone (XXIII), and of N-methyl-morphinane to give 3-hydroxy-N-methyl-morphinane (XXVIII).
2. Modifications of the N-containing part of the methadone molecule
Many compounds belonging to this class have been studied already by workers at Hoechst; their result that isomethadone (LVIII, in Chart IX) was relatively weakly active has more recently been contradicted.[45] Chen (loc. cit.) states that it is about half as potent as methadone. Eddy, Touchberry and Lieberman (loc. cit.) find it both less analgesic and less toxic than methadone, again the l-isomer being the active one; they state that parallel changes in the structures of methadone and isomethadone result in parallel modifications of activity.
Replacement of the -N(CH 3) 2 group in methadone and isomethadone by other nitrogen-containing residues has been extensively studied; especially morpholino- and piperidino- compounds have evoked much interest. The compound, d-l,-6-morpholino-4,4-diphenyl-3-heptanone CB 11, heptalgine, etc., has been placed on the market in England, and is stated to be significantly less toxic than methadone, with equal analgesic activity.[46] Claims for high activity and comparatively favourable toxicity have been put forward by Ofner, Thorp and Walton[47] for the piperidino-analogs of both methadone and isomethadone.

3. Modifications of the ketonic side-chain
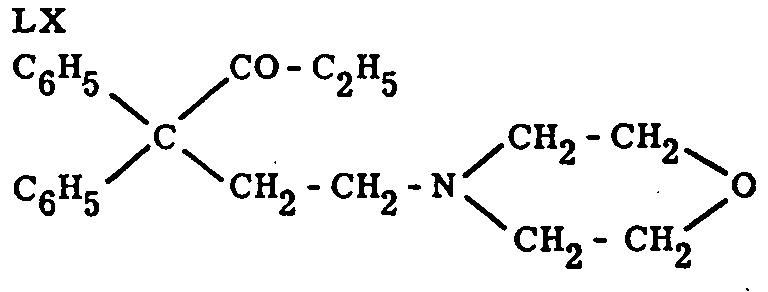
Replacement of the entire -CO-C 2H 5 chain by -O-COR in the analgetically active compound LX led to complete loss of activity, in contrast to the effect of the replacement of -COOR by -O-COR in pethidine.[48] Replacement of -CO-C 2H 5 by -COOR results in reduced activity (Eddy, Touchberry, Lieberman, loc. cit.).
Modifications of the carbonyl group alone point to a strong specificity. However, activity is retained in the remarkably stable ketimines* obtained as intermediates in the synthesis of methadone and its analogs; also the acetates of those imines are active analgesics.[49]
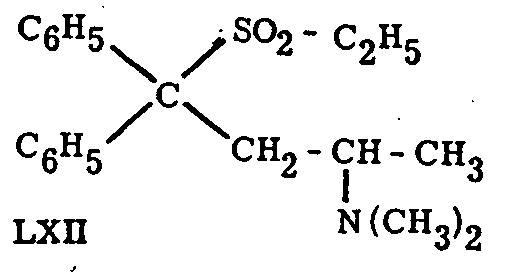
Replacement of the carbonyl by the sulfone-group equally leads to highly active analgesics.[50] Of the compounds prepared, Win 1161-2 (LXII) was the best one. It is claimed to combine the activity of methadone with greatly reduced toxicity. It has been resolved into optical antipodes51, of which the l-isomer is described as one of the most powerful analgesics.[52]
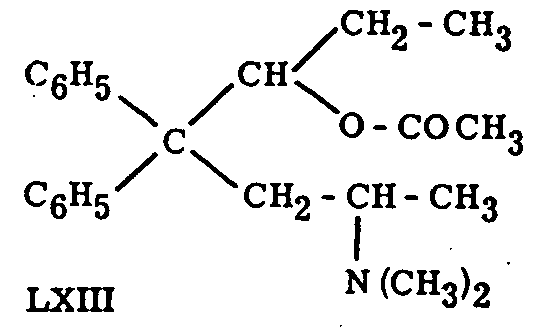
Reduction of the keto-group of methadone and isomethadone to a secondary hydroxyl has been studied in detail.53,54 The reduction succeeds in both cases with LiAlH 4, in the case of methadone also on catalytic hydrogenation. Only one of the two possible diastereomers was formed in either case. The resulting methadols and isomethadols were found to be only weakly active, but their esters are strongly analgetic (Eddy, Touchberry, Lieberman, loc. cit.); as a matter of fact, the one of the diastereomeric acetylmethadols (LXIII) resulting from reduction of l-methadone, followed by acetylation, is stated by Chen (loc. cit., p. 91) to be twice as active as d,l-methadone54, 55 while the diastereomer obtained in an analogous manner from d-methadone has created much interest through its prolonged activity.
One further group of compounds should be mentioned, which, although related to methadone, does not fit into any of the above sub-groups.
Adamson and Green[56] claim that compounds of the type LXIV are as active analgesics as morphine in the rat. The structural resemblance to methadone is unmistakable, but the absence of the quaternary carbon atom, present in nearly all other analgesics, makes this group of compounds theoretically interesting. The compound LXIV with R=R'=C 2H 5 has been designated as C-49.
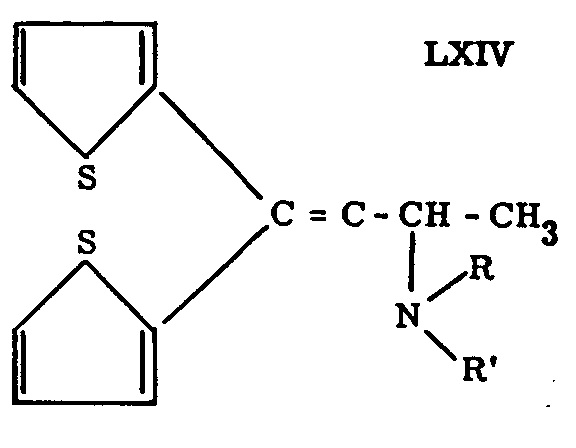
* See formula LXI, Chart IX.
It remains for us to mention a few of the many synthetic compounds which cannot be included in any one of the three preceding groups, and for which claims of analgesic activity have been made in published articles or patents. None of the numerous substances seem to have attained practical importance. For a more complete review we refer to the article by Bergel and Morrison loc. cit.
Many derivatives of 1, 2-diaryl-ethylamine have been prepared and tested. Analgesic activity had been originally claimed by Dodds et al[57] , but the claims have been disputed. More recently Holck, Kimura and Kimura[58] have investigated the activity, in mice, of 67 compounds of this group; they found that only one substance, one of the two diastereomers-of C 6H 5-CHOH-CH(NH 2)C 6H 5, had "fair" activity. Many more compounds of this type have been described by several groups of investigators.
Among a series of related tertiary amines prepared by Lee, Ziering, Berger and Heineman[59] the compound p-HOC 6H 4-CH 2CH 2-N(CH 3)-CH 2-CH 2-C 6H 5 had the highest potency (one-seventh that of morphine). Related compounds had been described earlier by Kuelz[60] .
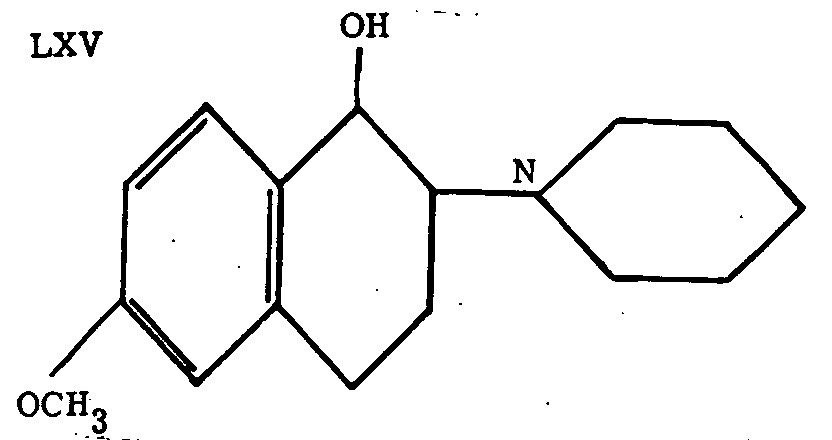
The compound LXV was described by Scheuing and Walach[61] as the most active of a number of related substances.
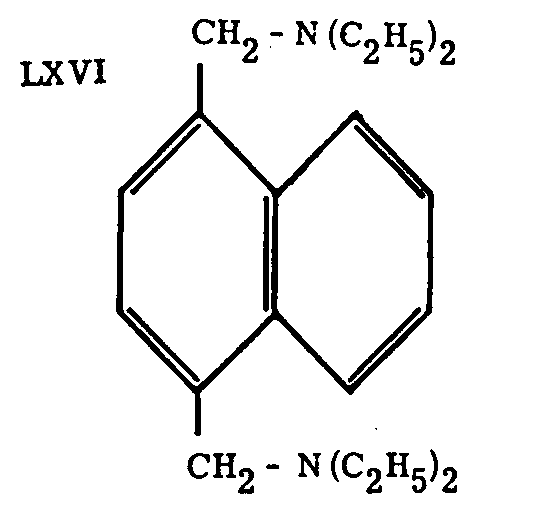
The compound LXVI is claimed by Badger, Cook, Donald, Graham and Walker[62] to be about as active as pethidine, which is remarkable in a compound not having any recognizable chemical relation to the morphine structure.
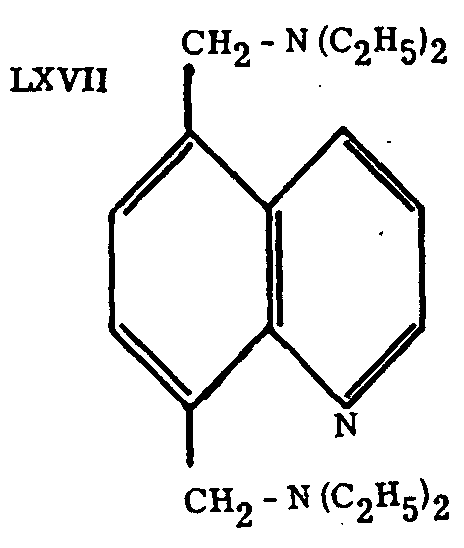
The closely related compound LXVII, prepared by Martin and Hanslick,[63] is described as a potent analgesic.
J. M. Gulland and R. Robinson, Mem. Proc. Manchester Lit. Phil. Soc. 69, 79 (1925)
2R. Grewe, A. Mondon and E. Nolte, Ann. 564, 161 (1949)
3L. F. Small, K. C. Yuen and L. K. Eilers, Journ. Amer. Chem. Soc. 55, 3863 (1933)
4L. F. Small, H. M. Fitch and W. E. Smith, Journ. Amer. Chem. Soc. 58, 1457 (1936)
5L. F. Small, Annals New York Acad. Sci., 51, 12 (1948)
6L. F. Small and H. Rapport, Journ. Org. Chem. 12, 284 (1917)
7National Research Council, Report of Committee on Drug Addiction (1929-1941)
8O. Eisleb and O. Schaumann, Deutsche Med. Woch., 65, 967 (1939)
9F. F. Yonkman, Annals New York Acad. Sci., 51, 59 (1948)
10F. Bergel and A. L. Morrison, Quart. Revs. (London) 2, 349 (1948)
11Emil Barell, Jubilee Volume
12N. B. Eddy, Journ. Amer. Pharm. Ass. 39, 245 (1950)
13P. O. Wolff, Bull. World Health Org., 2, 193 (1949)
14Annals New York Acad. Sci., 51, (1948)
15R. H. K. Foster and A. J. Carman, Journ. Pharm. and Exp. Therap. 91, 195 (1947)
16M. Bockmuehl and G. Ehrhart, German Patent 711069; C.A. 37, 4075 (1943)
17Grewe, Naturwissenschaften 33, 333 (1946); R. Grewe and A. Mondon, Chem. Ber. 81, 279 (1948)
18Journ. Chem. Soc., 1947, 399
19Journ. Org. Chem., 14, 1013 (1949)
20Journ. Org. Chem., 15, 1003 (1950)
21Journ. Amer. Chem. Soc., 71, 1359 (1949)
22Fieser and Holmes, Journ. Amer. Chem. Soc., 58, 2319 (1936); ibid. 60, 2548 (1938); Ghosh and Robinson, Journ. Chem. Soc. 1944, 506; Holmes and Mann, Journ. Amer. Chem. Soc., 69, 2000 (1947)
23Ber., 72, 785, 1314 (1939); 76, 1072, 1076 (1943)
24Helv. Chim. Acta, 32, 821 (1949)
25Gates and Newhall, Experiential, 5, 285 (1949); Gates, Woodward, Newhall and Kunzli, Journ. Amer. Chem. Soc., 72, 1141 (1950)
26Chemiker-Zeitung, 74, 314 (1950)
27Schnider and Gruessner, U.S.P. 2,524.855, C.A. 45, 1630a (1951); U.S.P. 2,524.856, C.A.45, 2030f (1951)
28Helv. Chim. Acta, 33, 1437 (1950)
29Journ. Amer. Chem. Soc., 72, 4839 (1950)
30Journ. Org. Chem. 3, 618 (1939)
31Office of the Publication Board, PB 981, 94; B.I.O.S., Final Report No. 766, items no. 22, 24, p. 60
32F. Bergel, A. L. Morrison and H. Rinderknecht, Journ. Chem. Soc. 1944, 265
33E. Walton and M. B. Green, Journ. Chem. Soc., 1945, 315
34Brit. Pat. 591.992, C.A., 42, 1322 (1948)
35W. Tarpey, H. Hauptmann, B. M. Tolbert and H. Rapoport, Journ. Amer. Chem. Soc. 72, 5126 (1950).
36O. Schaumann, Arch. Exp. Path. Pharm., 196, 109 (1940)
37O. Eisleb, Ber. 74, 1433 (1941)
38Kleiderer, Rice and Conquest, Office of the Publication Board, P.B., 981, 96; B.I.O.S., Final Report No. 116, item, 24, p. 51
39Small, Annals New York Acad. Sci., 51, pp. 14 ff. (1948); Easton, Gardner and Stevens, Journ. Amer. Chem. Soc., 69, 2941 (1947); Schultz and Sprague, ibid., 70, 48 (1948); Schultz, Robb and Sprague, ibid., 69, 188, 2454 (1947); Brode and Hill, ibid., 69, 724 (1947)
40Nature, 159, 679 (1947); 160, 605 (1947)
41Journ. Org. Chem., 13, 191 (1948)
42Journ. Amer. Chem. Soc., 70, 4194 (1948)
43Journ. Amer. Chem. Soc., 71. 2935 (1949)
44Literature quoted in Chen, loc. cit., p. 90; Denton and Beecher, Journ. Amer. Med. Assoc., 141, 1146 (1949)
45Luduena, Miller, Ananenko and Frick, Federation Proc. 7, 241 (1948)
46Wilson and Hunter, Brit. Med. Journ. 1948, 553; Speeter, Byrd, Cheney and Binkley, Journ. Amer. Chem. Soc., 71, 57 (1949); literature: Basil, Edge and Somers , Brit. Journ. Pharmacol., 5, 125 (1950); Winter and Flataker, Journ. Pharmacol. Exper. Therap., 98, 305 (1950)
47Nature, 163, 479 (1949)
48Beckett and Linnell, Journ. Pharmacy and Pharmacology, 2, 427 (1950)
49Cheney, Smith and Binkley, Journ. Amer. Chem. Soc., 71, 53 (1949)
50Klenk, Suter and Archer, Journ. Amer. Chem. Soc., 70, 3846 (1948)
51Tullar, Wetterau and Archer, Journ. Amer. Chem. Soc. 70, 3959 (1948)
52Lewis, Journ. Pharmacol. Exp. Therap. 96, 31 (1949)
53May and Mosettig, Journ. Org. Chem., 13, 459, 663 (1949); Speeter, Byrd, Cheney and Binkley, Journ. Amer. Chem. Soc. 71, 57 (1949)
54cf. also: Sherrod, Kaiser, Santos-Martinez and Pfeiffer, Federation Proc., 7, 255 (1948)
55Chen, loc. cit., p. 91; Pohland, Marshall and Carney, Journ. Amer. Chem. Soc., 71, 460 (1949)
56Nature, 165, 122 (1950)
57Nature, 151, 614 (1943) ; 154, 514 (1944); Proceedings of the Royal Society, B 132, 119 (1944) ; Journ. Physiol. 104, 47 (1945)
58Journ. Amer. Pharm. Assoc., 39, 354 (1950)
59Emil Barell, Jubilee Volume 1946, p. 264
60U.S.P. 2,276,618; 2,276,619; C.A., 36, 4672 (1942)
61U.S.P. 2,369,611; C.A. 39, 5042 (1945); 2,352,020; C.A. 38, 5645 (1944)
62Nature, 162, 21 (1948)
63U.S.P. 2, 507,313; CA. 44, 8381a (1950)


