EXPERIMENTAL
Analytical procedures
THEORY
EXPERIMENTAL RESULTS AND DISCUSSION
SUMMARY AND EVALUATION OF THE METHOD
ACKNOWLEDGEMENTS
Author: Leo Levi, P.M. Oestreicher, Charles G. Farmilo
Pages: 15 to 25
Creation Date: 1953/01/01
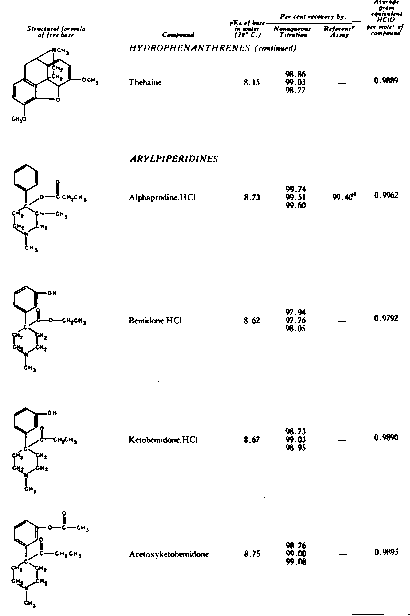
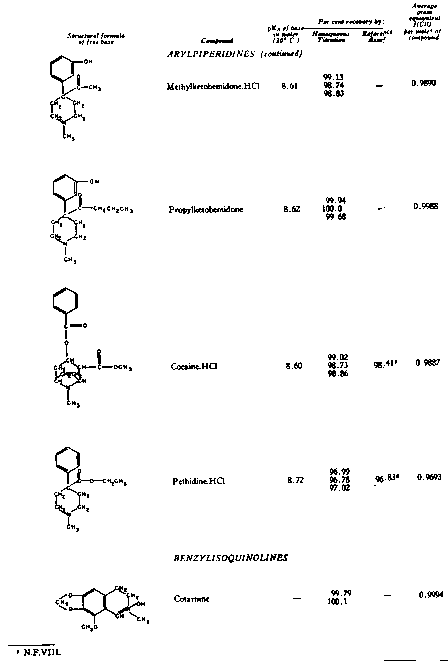
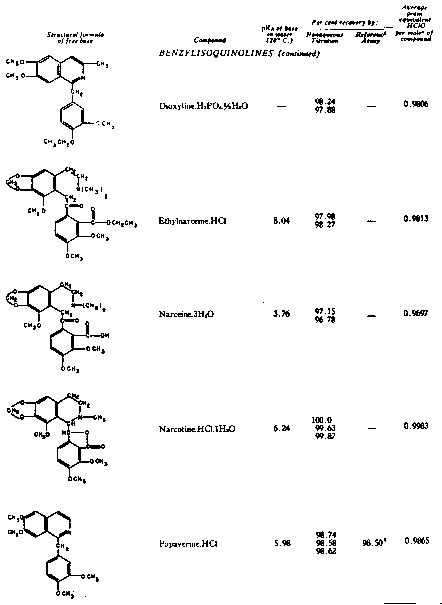
Twenty-five years ago, Conant, Hall, and Werner showed that many substances which exhibit little or no basic properties in water behave as relatively strong bases in glacial acetic acid and may be quantitatively determined in this solvent by titration with a strong mineral acid ([1] ), [(2)] .2 Unfortunately, the potentialities of this method and its distinct advantages, as compared to many volumetric procedures carried out in aqueous media, were not immediately realized. It is only within the last two years that nonaqueous titrimetry has found a considerable number of novel applications and become a valuable analytical method of wide scope [(3)] , [(4)] . The studies reported in this paper demonstrate that the quantitative estimation of narcotic drugs and alkaloids of widely different structures and degrees of basicity may also be achieved by means of nonaqueous titration, using acetic acid as solvent and perchloric acid as titrant.
Reagents and solutions
Perchloric acid 0.05 N in glacial acetic acid, prepared by dissolving approximately 4.2 ml. of perchloric acid 70-72 per cent A.C.S. grade in 1 liter of glacial acetic acid containing 25 ml. of acetic anhydride to react with any water introduced into the system (perchloric acid, water of crystallization and moisture of the sample). Standardization against N.B.S. potassium acid phthalate was performed after 24 hours standing in accordance with the procedure given by Seaman and Allen [(5)] .
Acetic anhydride, A.C.S. grade.
Acetic acid glacial, A.C.S. grade.
Mercuric acetate solution, made up by dissolving 6 gm. of mercuric acetate C.P. in 100 ml. of hot glacial acetic acid and allowing to come to room temperature.
Methyl violet (6B) indicator solution, prepared by dissolving 0 1 gm. of dye in 100 ml. of glacial acetic acid.
11. Contribution from the Organic Chemistry and Narcotic Section, Food and Drug Laboratories, Department of National Health and Welfare, Ottawa, Canada
22. Figures in parentheses indicate items within the list of references to be found at the end of this article.
All compounds except the halide acid salts were analysed by dissolving about 0.5 milliequivalents in 80 ml. of glacial acetic acid and titrating directly with 0.05 N perchloric acid to a blue-green end point. A microburet calibrated to 0.01 ml. was used for measuring the volume of acid consumed.
The halide acid salts were dissolved in 80 ml. of glacial acetic acid, reacted with 5 ml. of 6 per cent mercuric acetate solution and subsequently neutralized by means of perchloric acid as indicated above. A magnetic stirrer was used to carry out the determinations more conveniently.
Acetic acid is generally considered an amphiprotic solvent for it can act as both a proton donor and acceptor. The dissociation constant of its autoprotolysis which proceeds in accordance with the equation:
2CH 3COOH === CH 3COOH 2+ + CH 3COO- was found by Kolthoff to be equal to 2.5X10-13 at 25°C. (6).
When a base B is dissolved in acetic acid the ionization process:
B + CH 3COOH BH + + CH 3COO -
takes place to an appreciable extent and even with bases that are considered weak in aqueous media the equilibrium of the reaction lies far to the right. This "levelling effect" of the solvent was first observed by Hall who showed by means of electrochemical measurements that all bases stronger than aniline, which is a very weak base in water (pK A=4.75), are of considerable and almost equal strength in acetic acid solution [(7)] , [(8)] . It is evident that in these reactions the solvent displays strong protogenic or acidic properties.
Addition of perchloric acid to acetic acid is associated with the formation of a solvated proton:
HClO 4 + CH 3COOH ==== CH 3COOH 2+ + ClO 4-
and under normal conditions this reaction tends to go to completion [(6)] , [(9)] . Hence, in a perchloric acidacetic acid mixture, acetic acid displays protophilic or basic properties. The solvated ion generated is a powerful proton donor, i.e., an acid of exceptional strength. It will readily neutralize any base that is stronger than the solvent itself and the fundamental process underlying nonaqueous titration of bases in glacial acetic acid by means of perchloric acid may thus be visualized to proceed in the following manner:
B + CH 3COOH === BH+ + CH 3COO-
CH 3COO- + CH 3COOH 2+ + ClO 4 - =
2 CH 3COOH + C10 4 -
Salts of organic bases - except sulfates and halides - are neutralized in a similar fashion as may be shown for codeine phosphate:
C 18H 21NO 3 H 3PO 4=== C 18H 21NO 3H+ + H 2PO 4-
H 2PO 4- + CH 3COOH 2 + = H 3PO 4 + CH 3COOH
Sulfates are titrated to bisulfates only because the in the reaction
SO 4-- + H+ (acetic acid) HSO 4- the equilibrium lies far to the right whereas in the succeeding reaction
HSO 4- + H+ (acetic acid) H 2SO 4 it lies far to the left. Bisulfates therefore do not behave as bases in glacial acetic acid (10).
Halide ions are extremely weak bases and the reaction
X - + CH 3COOH 2 + === HX + CH 3COOH
does not go to completion, the strong mineral acid generated during the titration causing the end point to occur long before the equivalence point is reached.
Correct results can be obtained however by either boiling the solution to volatilize the hydrogen halide formed and thereby force the reaction to completion - a method analogous to the titration of carbonates in aqueous solution - or by adding to the system prior to titration a slight excess of mercuric acetate so as to convert the anion to undissociated mercuric halide which will not take part in the neutralization process [(l1)] , [(12)] . The latter procedure is to be preferred because of the tendency of many organic compounds to decompose on prolonged boiling. The reaction appears to proceed in accordance with the following scheme:
R 3N. HX + 1/2 Hg (CH 3COO) 2 =
R 3NH+ + CH 3COO- + ? HgX 2
CH 3COO + CH 3COOH 2 + = 2 CH 3COOH
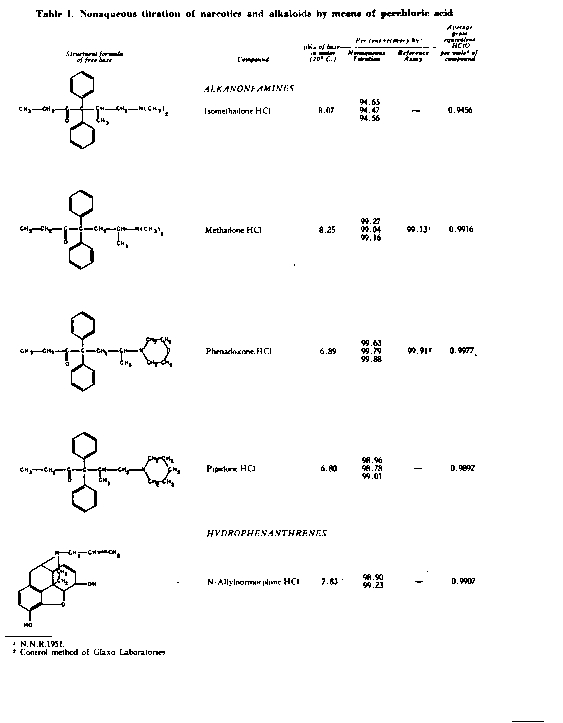
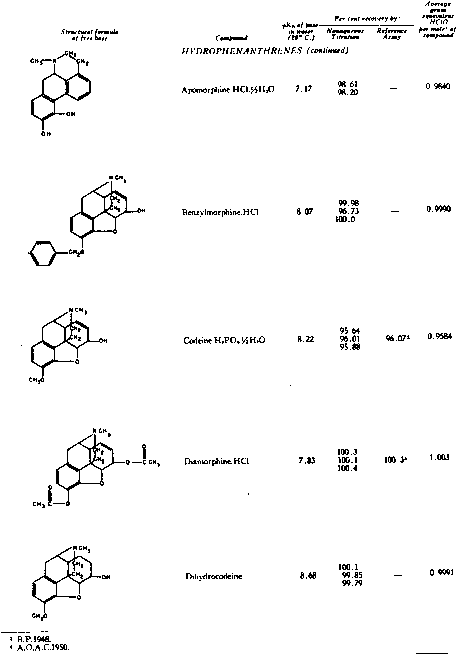
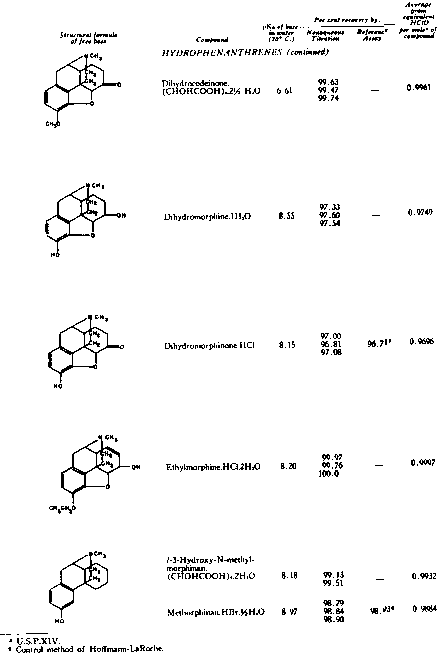
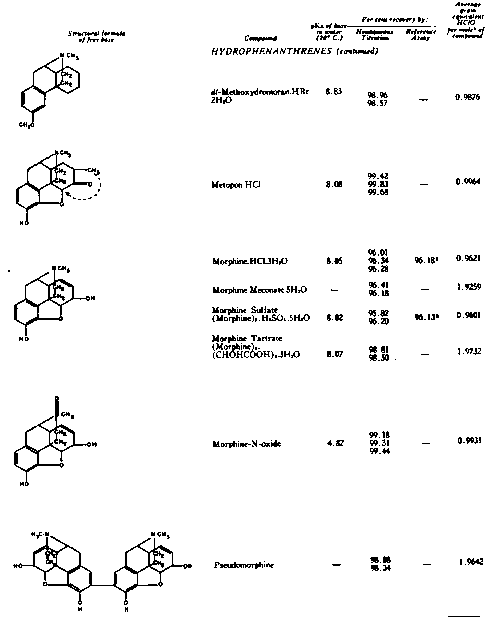
The results of the analyses presented in table I illustrate that a variety of narcotics and alkaloids may be accurately determined in glacial acetic acid by titration with perchloric acid. The compounds investigated comprise diarylalkoneamines, arylpiperidines, benzylisoquinolines, and hydrophenanthrenes containing an imino-ethano linkage. These amines are only slightly soluble in water and because of their weakly basic character most of them cannot be determined in aqueous media with a high degree of precision. The structural formulae of the organic bases are shown in column 1 and their aqueous dissociation constant exponents, determined in accordance with the method of Saunders, given in column 3 [(13)] . Papaverine, phenadoxone, pipidone, and narcotine, for example, with pK A values less than 7, did not show any inflection in their titration curves when water was used as solvent and nonaqueous titrimetry offers a valuable direct method for their quantitative determination. In accordance with the theoretical considerations given morphine sulphate used but one mole of perchloric acid for complete neutralization:
(C 17H 19NO 3 H) 2 SO 4 + CH 3COOH 2+ =
2 C 17H 19NO 3H+ + HSO 4- + CH 3COOH
whereas morphine tartrate consumed two moles of titrant per mole - see column 5 - because the tartrate ion which is neutralized carries two negative charges:
(MH) 2++ + CHOHCOO- + 2 C10 4- +2 CH 3COOH 2+ =
CHOHCOO-
(MH) 2++ + 2 C10 4- + (CHOHCOOH) 2 + 2 CH 3COOH
Dihydrocodeinone and dromoran ( 1-3-hydroxy-N-methyl morphinan) tartrate however consumed only one mole of perchloric acid per mole because the tartrate ion which is titrated in these instances carries but one negative charge:
BH+ + CHOHCOO- + C10 4- + CH 3COOH 2+ =
CHOHCOO-
BH+ + C10 4- + (CHOHCOOH) 2 + CH 3COOH
Pseudomorphine was found to consume two equivalents of perchloric acid per mole as expected because two acetate ions are affected by the titration. In column 6 results of analyses using official and industrial control methods are given and it is evident that good agreement exists with the results obtained by titration with perchloric acid (column 4).
The quantitative estimation of morphine-N-oxide by means of perchloric acid is of particular interest since it represents - to the best of our knowledge - the first reported nonaqueous titration of an amine oxide. It is to be realized that, whereas the basicity of an amine is determined by the reactivity of the unshared pair of electrons associated with the nitrogen atom, the basic character of an amine oxide is a function of the oxygen rather than of the nitrogen atom for there is on the latter no lone electron pair to which a proton could become attached. Hence formation of the perchlorate of an amine oxide in acetic acid is comparable to the formation of its halide acid salt in water. However, most amine oxides are not sufficiently basic to be titratable with halide acids in aqueous media. With morphine-N-oxide for example no potential break whatever can be detected during titration in water with a 0.05 N aqueous hydrochloric acid solution whereas titration in glacial acetic acid with a perchloric acid solution of comparable strength is associated with a distinct colour change at the equivalence point. Since tribenzylamine oxide can also be determined by means of perchloric acid [(14)] it may well be that amine oxides in general fall within the scope of this method and that other heterocyclic compounds whose basicity and capability of salt formation are due to the presence of unshared pairs of electrons associated with an oxygen atom, e.g., pyrones and related compounds may also be estimated by titrimetry in nonaqueous solvents
In addition to its application to pure compounds, as shown in table I, the method has been successfully used for some time in this laboratory for the rapid and accurate analysis of narcotic seizures. Non-ionizing adulterants such as starch, sucrose, lactose, aspirin, etc., do not show any interference but since anions react with perchloric acid an excipient like calcium carbonate for example would affect the titration and should be removed from the sample in order to avoid erroneous titration values The presence or absence of nitrogen containing diluents, such as quinine, novocaine, and amidone, which are occasionally encountered in illicit samples must, of necessity, also be established and corrected for prior to analysis.
In all titrations vivid colour changes accompanied the approach of the equivalence point and two or three drops (approximately 0.03 ml.) of the titrant were always sufficient to change the colour of the solution from the last shade of blue to a faint green.
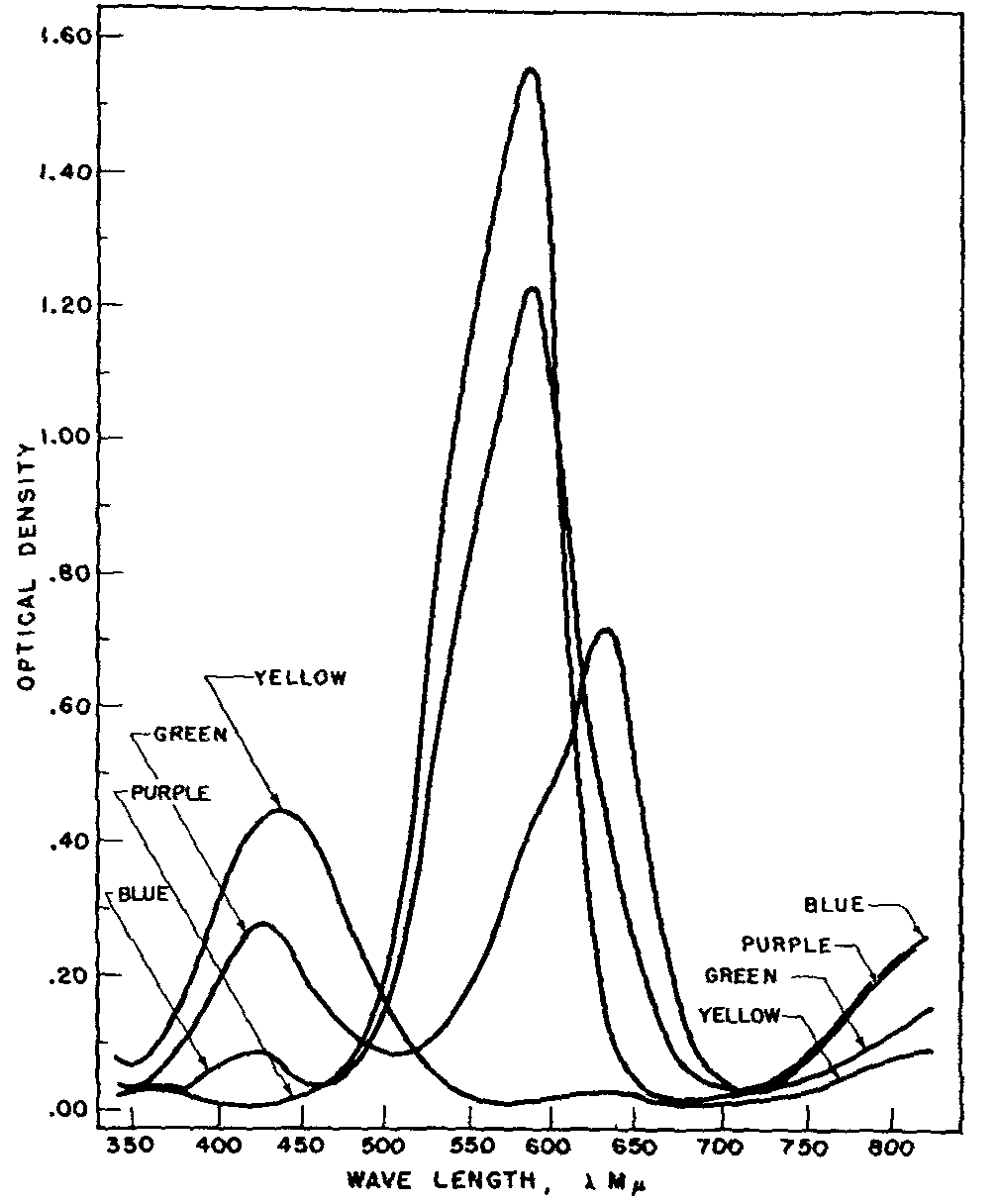
Methyl violet, the indicator used, shows a variety of colours in glacial acetic acid depending on the pH of the solution. These colours were observed by titrating 30 ml. of a solution containing 3.8x10 -7 moles of methyl violet (pentamethyl-p-rosaniline hydrochloride) per litre with 0.009196 N perchloric acid. It was found that 0.110 ml. of the acid changed the colour of the indicator from purple to blue, 0.161 ml. from blue to green and 1.650 ml. from green to yellow.
The spectral curves shown in figure 1 illustrate the absorption of the indicator at these three levels of perchloric acid titration. They resemble those obtained by Conant and Werner on crystal violet (hexamethyl-p-rosaniline hydrochloride) in buffered glacial acetic acid systems but show that additional maxima and another isobestic point occur below 475 m? which region these workers had left unexplored [(15)] . Since the addition of perchloric acid to a solution of methyl violet in glacial acetic acid is associated with progressive protonation of the indicator the isobestic points occurring at about 350, 500 and 725 m? may be visualized as corresponding to formation of the three ionic species:

Some of the compounds assayed, e.g., dihydrocodeinone, formed perchlorates which were only partially soluble in the solvent system used and precipitated during the titration. However this phenomenon did not reduce the sharpness of the indicator end point. Solutions of apomorphine in glacial acetic acid developed a blue coloration on standing and had to be titrated fairly rapidly to obtain accurate assay values. It was also found important to carry out all determinations under essentially anhydrous conditions for the sharpness of the indicator end point was noticeably impaired by the presence of about 2 per cent of water, and the volume of titrant consumed in such cases exceeded always the stoichiometrically required amount - apparently because of the weakly basic properties inherently exhibited by water:
CH 3COOH 2+ + H 2O === CH 3COOH + H 3O +
The use of a slight excess of acetic anhydride helped prevent such interference. Another factor that had to be taken into account is the rather large coefficient of cubical expansion of glacial acetic acid which amounts to 0.11 per cent per °C [(5)] . In order to obtain accurate results the titrant was therefore always standardized and used at the same temperature. Provided these precautions were met replicate determinations agreed very closely and a precision of ±0.3 per cent was easily realized. This value compares favourably with that obtained by conventional aqueous acidimetric and alkalimetric titrations.
The data presented show that nonaqueous titrimetry permits the rapid and accurate direct determination of narcotic bases and, inter alia, the quantitative estimation of the physiologically active moiety of the molecule if it occurs as the salt of an organic or inorganic acid. The apparatus needed to carry out these analyses is inexpensive and available in any laboratory equipped to do aqueous titrations. The method itself is free from all processes of time-consuming extractions, chromatographic separations, formation of insoluble derivatives under controlled conditions, and reference to colorimetric standards, which techniques characterize most official and industrial procedures. It should prove of particular value to toxicologists and forensic chemists concerned with the quantitative evaluation of these drugs.
The authors wish to express their appreciation to Dr L. I. Pugsley, Chief, Food and Drug Laboratories, for his valuable advice and suggestions throughout the course of these studies
Conant, J B. and Hall, N. F., J. Am Chem Soc, 49 , 3047 (1927).
002Riddick, J R, Anal. Chem , 24, 41 (1952).
003Fritz, J. S, . Acid-Base Titrations in Nonaqueous Solvents , The G Frederick Smith Chemical Company, Columbus,Ohio (1952).
004Seaman, W and Allen, E., Anal. Chem , 23, 592 (1951)
005Kolthoff, I M. and Willman, A., J Am Chem Soc, 56 , 1007 (1934).
006Hantzsch, A, Zeitschr Physik Chem, 134, 406 (1928)
007Hall, N. F, Chem Reviews, 8, 191 (1931); J. Am Chem Soc, 52 , 5115 (1930).
008MacKenzie, H A. E and Winter, E. R S., Trans Farad Soc, 44 , 159 (1948).
009Pifer, Ch W and Wollish, E. G., Anal Chem, 24 , 519 (1952).
010Higuchi, I. and Concha, J, Science, 113 , 210 (1951)
011Pifer, Ch. W and Wollish, E. G, Anal Chem, 24 , 300 (1952)
012Hall, N F and Werner, T. H., J Am Chem Soc, 50, 2367 (1928).
013Davis, M. M, National Bureau of Standards, Washington, D C, Private Communication
014Saunders, L. and Srivastava, R. S, Journ. Pharm and Pharmac, 3, 78 (1951).
015Conant, J. B, and Werner, T. H, J. Am. Chem. Soc, 52, 4436 (1930).
They also gratefully acknowledge the contribution of supplies of materials used in this study by Bilhuber-Knoll Corp, Orange, N J, USA; Burroughs Wellcome and Company, London, England; Ciba Ltd., Basle, Switzerland; El[?] Lilly and Company, Indianapolis, Ind., U S A; Endo Products Inc,Richmond Hill, N Y, U S A.; Glaxo Laboratories Ltd, Greenford, Middlesex, England, Hoffmann-LaRoche Inc, Nutley, N.J, U S A; May and Baker Ltd, Montreal, Canada; Merck and Company Inc, Rahway, N.J, U S A.; Parke, Davis and Company, Walkerville, Ontario, Canada; T. and H Smith Ltd, Edinburgh, Scotland; Winthrop-Stearns Inc., Windsor, Ontario, Canada.