ORIGINAL GERMAN PROCESS
SUMMARY
VARIATIONS ON THE GERMAN PROCESS
OTHER METHADONE PROCESSES
RESOLUTIONS
REACTIONS OF METHADONE (16), (32)
REACTION'S OF ISOMETHADONE (37)
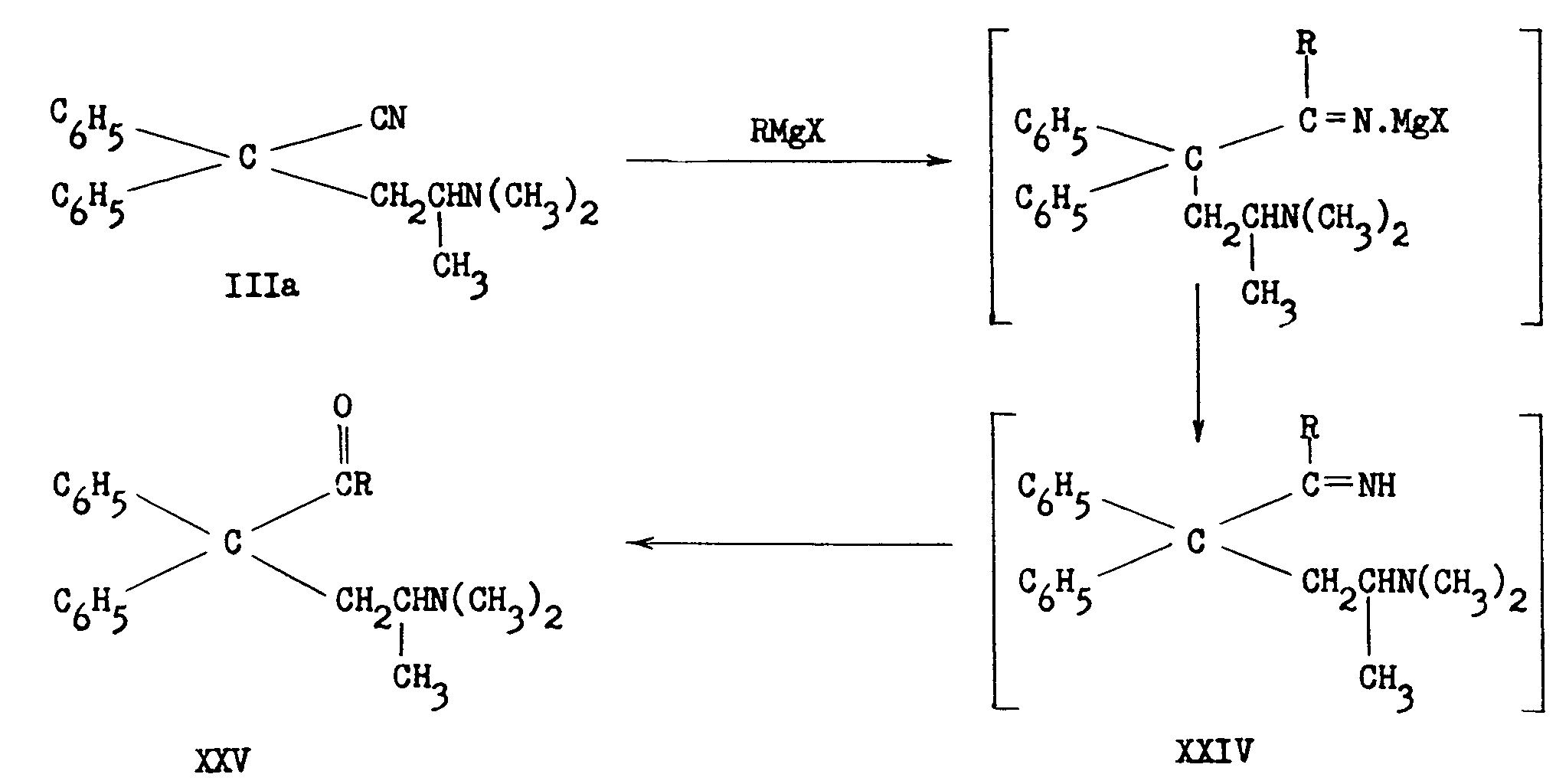
REACTIONS OF THE NITRILES IIIa AND IIIb
Author: Manuel M. Baizer
Pages: 32 to 43
Creation Date: 1953/01/01
In 1945 the United States Department of Commerce published[1] 1 the details of the process developed at the I.G. Farbenindustrie, Höchst am Main, for preparing the new potent morphine-like analgesic Methadone.2 Since that time a considerable body of chemical literature has appeared dealing with the elucidation of the original process. The development of modified processes, the separation of position and optical isomers of Methadone, the formation of derivatives and transformation products and the synthesis of numerous analogues.
It is the purpose of this paper to review the literature that has appeared up to the end of 1951 on methadone chemistry. Brief reviews of this subject appeared in 1948 [3] [4] .
Diphenylacetonitrile (I) 3 was condensed in the presence of sodium amide with IIA, the chloroamine (B.P. 60-63o at 110 mm.; stated to be 1-dimethyl-amino-2-chloropropane) obtained by the action of thionyl chloride on 1-dimethyl-amino-2-propanol. The amino-nitrile product (III), obtained as a partially crystalline material, was brought into reaction with slightly more than two moles of ethylmagnesium bromide. The Grignard reaction product was decomposed while hot with hydrochloric acid whereupon crude Methadone 4 hydrobromide (IVA. HBr) precipitated (yield, 49.7 per cent of theory based upon III). The hydrobromide was converted to free base IVA and then to methadone hydrochloride.
1Arabic figures in parentheses indicate items within the list of references to be found at the end of this article.
2 This generic name, accepted by the Council on Pharmacy and Medicine of the American Medical Association ([2] ) will be used throughout this review to denote 4,4-diphenyl-6-dimethyl-amino-3-heptanone This substance is also known by the following names, some of which are proprietary trade-marks Adanon, Amidone, Amidosan, AN-148, Butalgin, Depridol, Diaminon, Dianone, Dolafin, Dolamid, Dolophine, Dorexol, Heptadon, Heptanal, Hoechst 10820, Ketalgm, Mecodm, Mephenon, Miadone, Moheptan, Physeptone, Physopeptone, Polamidon, Symoran, Turanone.
3 Roman numerals indicate reference to structures illustrated within this article.
4 Since the molecule contains one asymetric carbon atom it is obtained in this synthesis in its d1-form.
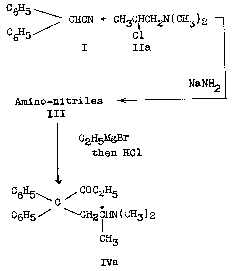
Kleiderer [5] pointed out that there was an apparent discrepancy between the structure of Methadone as represented in IVa and the one which might have been expected to result from the sequence of reactions described above, namely,
That the structure of Methadone was, however, truly represented by IVa was proved in several ways:
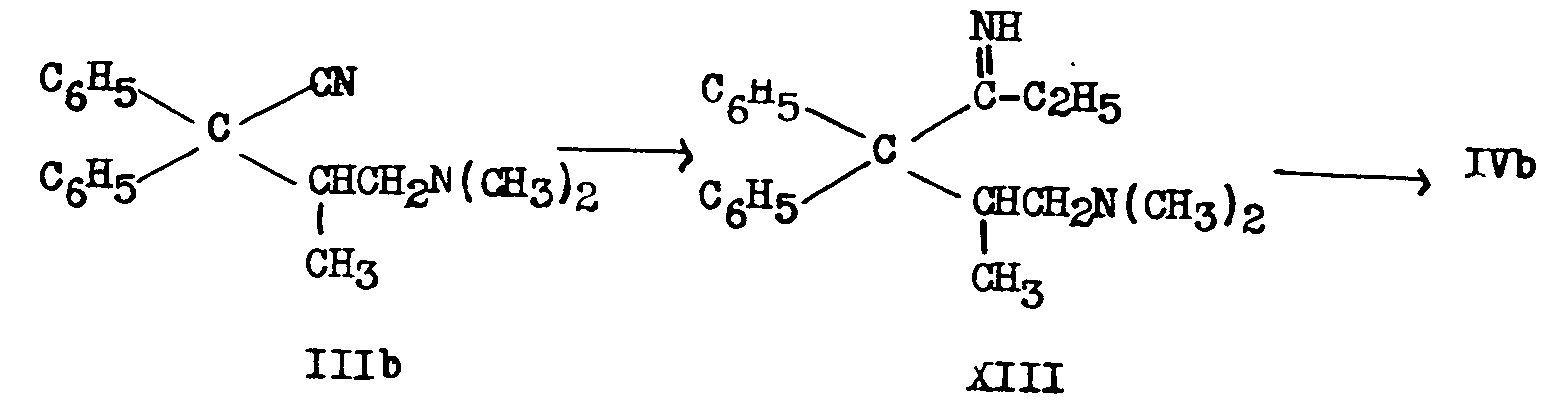
Schultz, Robb and Sprague [6] showed that III obtained in the German process consisted of about equal parts of 2-dimethylamino-4,4-diphenyl-valeronitrile (IIIa) and 1-dimethylamino-2-methyl-3,3-diphenyl-butyronitrile (IIIb) and that IIIa led to Metha-done. They proved the structure of each of the 2 amino-nitriles by the following sequence of reactions:
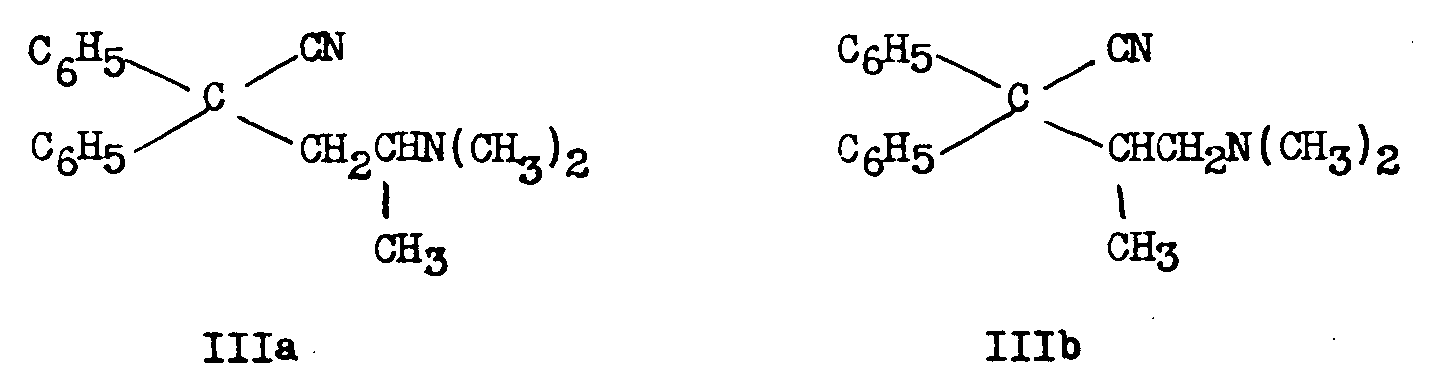
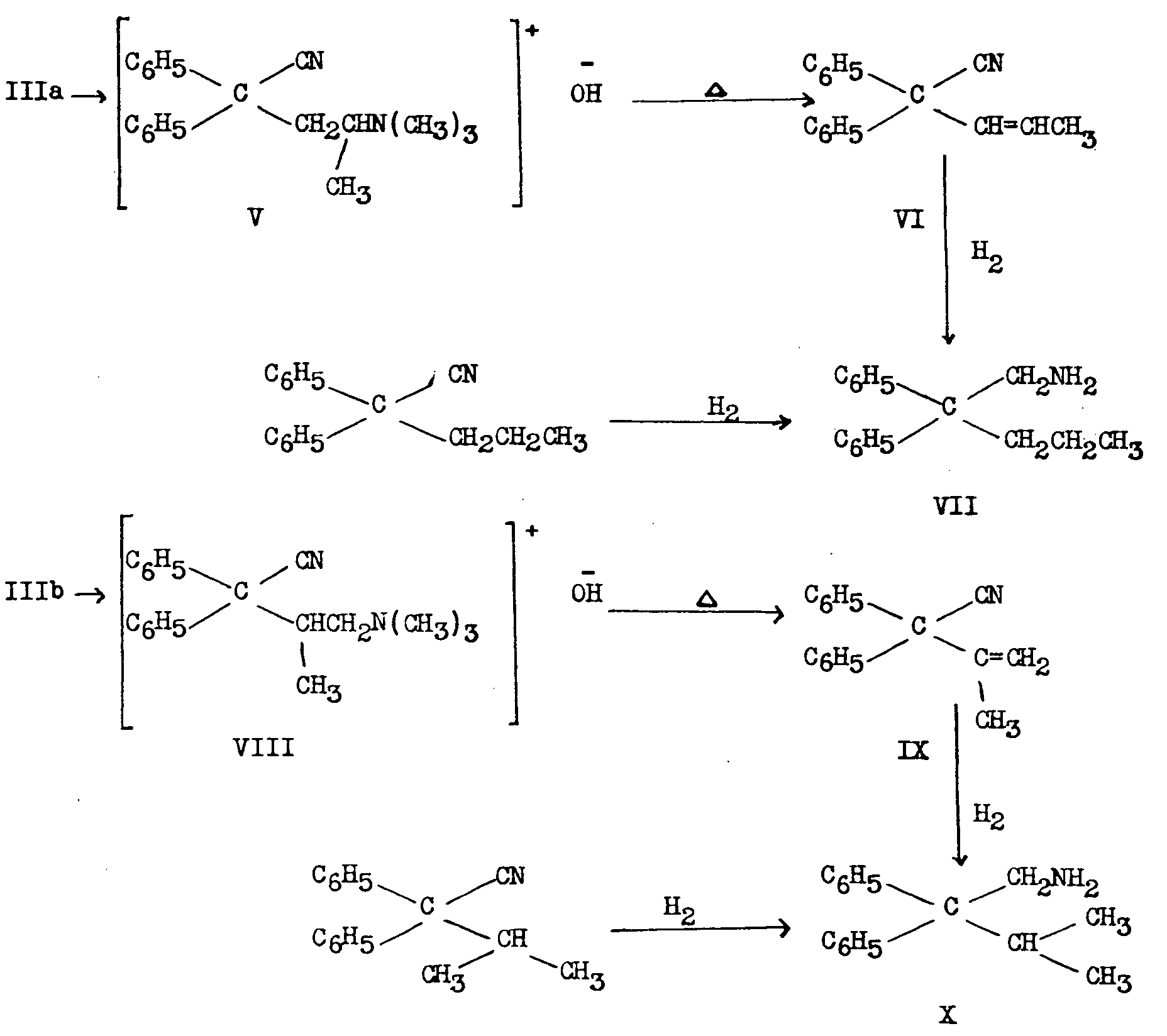
2. Bockmühl (7, 36) degraded IIIa and IIIb to 2-dimethylamino-4,4-diphenylbutane (XI) and 1-dimethylamino-2-methyl-3,3-diphenylpropane (XII) respectively and synthesized the latter two compounds by independant means.
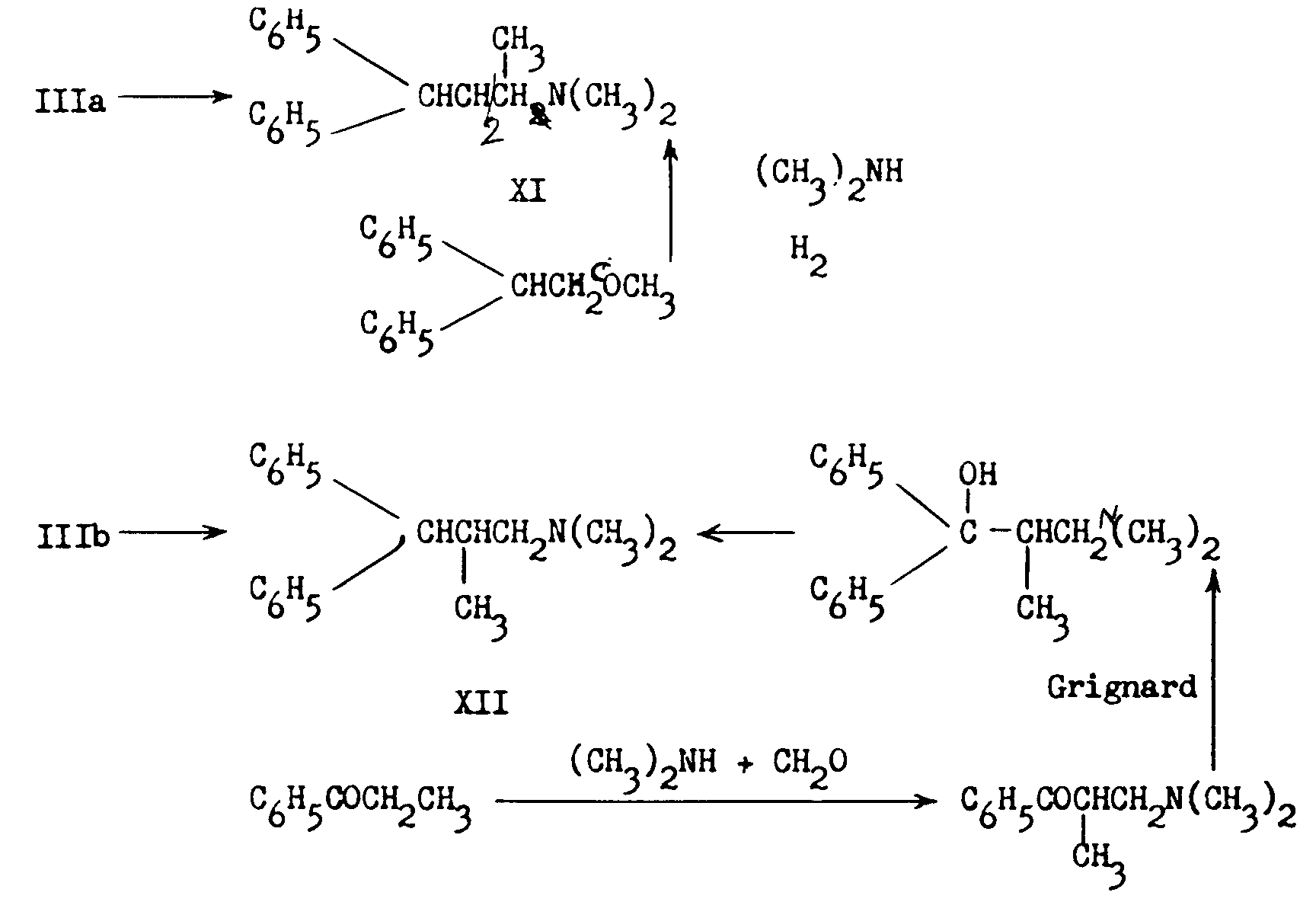
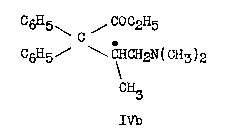
3. Easton, Gardner and Stevens' work ([8] ) confirmed the above results and in addition demonstrated5 that the reaction of ethylmagnesium bromide with IIIb led to the unusually stable ketimine (XIII), which on prolonged hydrolysis yielded isomethadone (IVb):
XIII and to a smaller extent IVb were present in the Mother liquors obtained after removal of Methadone hydrobromide in the original German process. The above authors verified the structure of Methadone in a novel manner:
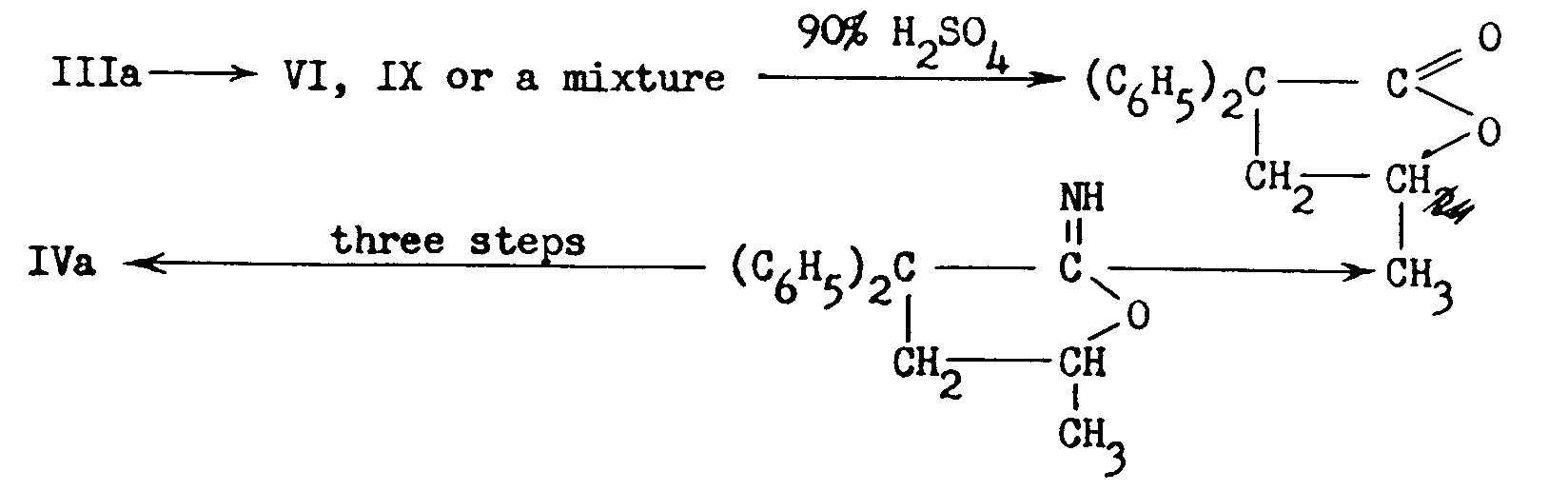
5Apparently anticipated by Slezinger and Tishler ([9] )
The mechanism of the formation of two6 isomeric nitriles (IIIa and IIIb) in the condensation of diphenylacetonitrile has been the subject of several inquiries. There was general acceptance of the proposal [6] , [7] that a cyclic ammonium ion was probably involved which could cleave in either of two ways and thereby give rise to two products:
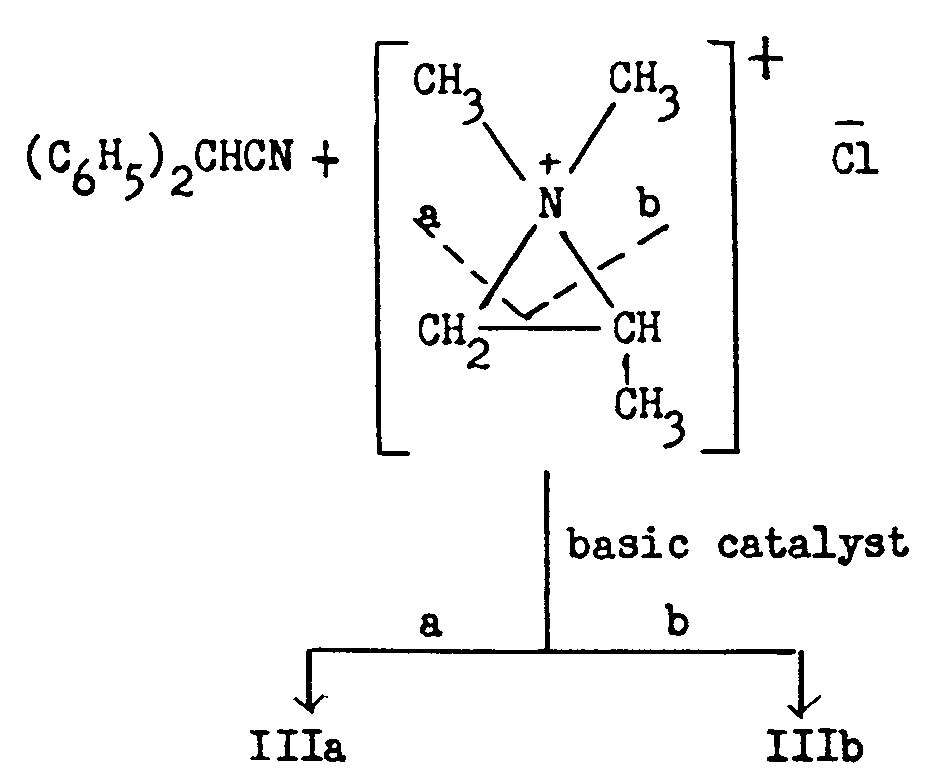
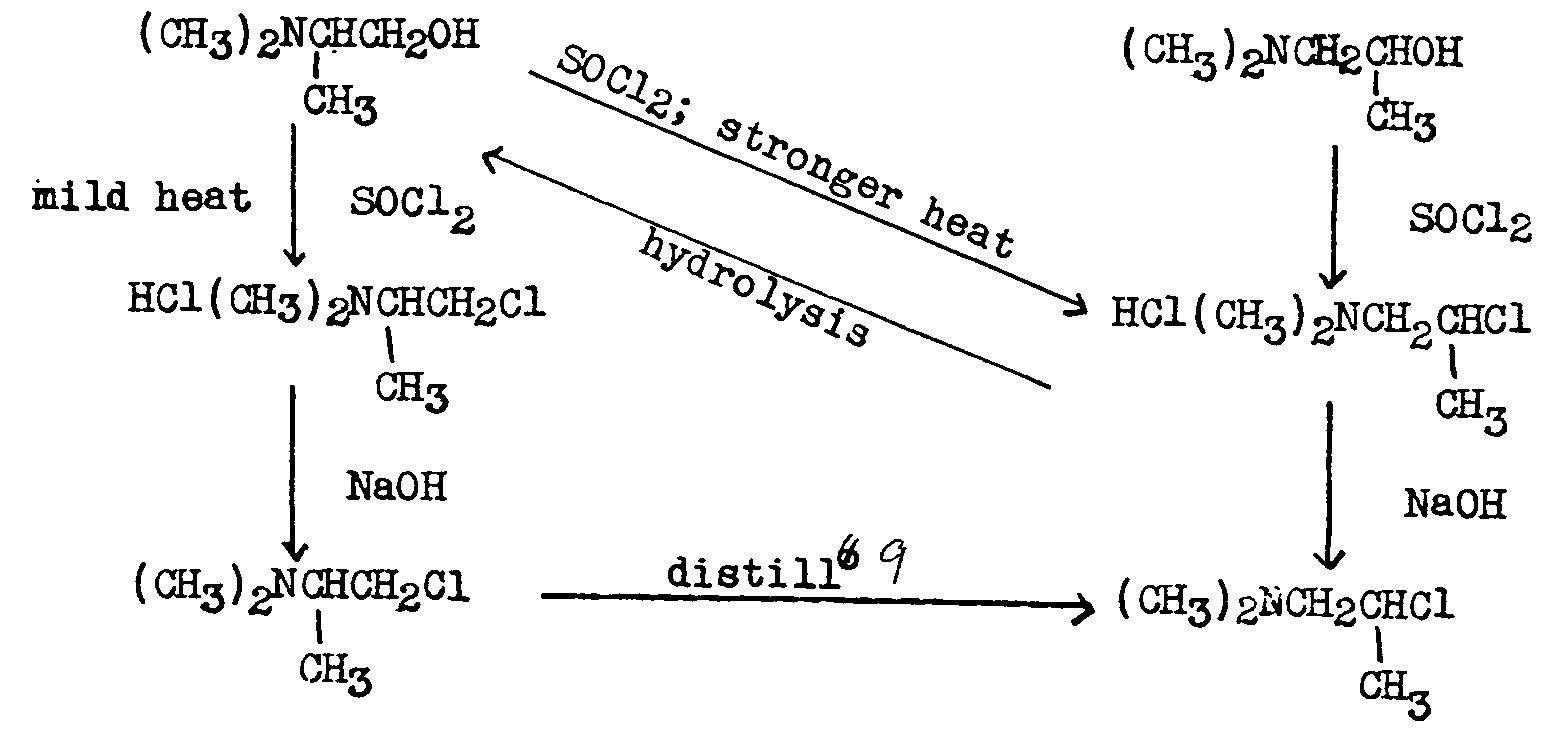
The point of disagreement was the interpretation of whether the cyclic ion was formed and cleaved before the condensation reaction or in the course of the condensation reaction. If the former view were correct, 1-dimethylamino-2-chloropropane (IIa) and 1-chloro-2-dimethylaminopropane (IIb) would be interconvertible and the chloroamine used in the German process would actually be an equilibrium mixture of these two isomers. 7
Schultz and Sprague ([11] ) reported that 1-dimethylamino-2-chloropropane (IIa) was thermally stable but that 2-dimethylamino-1-chloropropane (IIb) rearranges to IIa on heating and that IIb hydrochloride rearranges to IIa hydrochloride. Undistilled IIb liberated at a low temperature from its hydrochloride and condensed with diphenylacetonitrile yielded a mixture of the isomeric nitriles IIIa and IIIb. These authors concluded therefore that arrangement occurs in the course of the condensation of the chloroamine with the diphenylacetonitrile anion (basic catalyst present).
6 A third structural isomer of Methadone presumably arising from a third isomeric nitrile has been reported ([8b] ) but not verified by other invesitgators.
7 The ratio of the two isomers might then vary according to the conditions used in the liberation of the chloroamine from its hydrohalide salt. Cf.[10] .
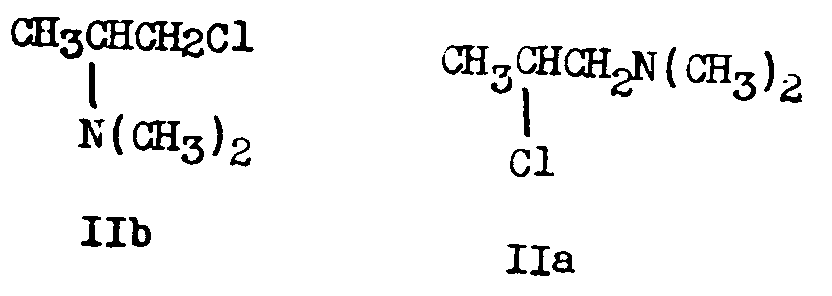
Walton, Ofner and Thorp ([12] ) confirmed the above work, but Brode and Hill ([13] ) held that both IIa and IIb rearranged, under the conditions used to liberate them from their respective hydrochlorides, to an equilibrium mixture of the two. Attenburrow, et al., ([14] ) found that IIb was not thermally unstable and did not isomerize during distillation.
The entire question of the isomerism of the chloroamines was later re-examined by Ofner ([10] ) whose results are summarized in the chart below.8
Ofner concluded that the mixture of amino-nitriles (IIIa and IIIb) obtained in the reaction between diphenylacetonitrile and either 1-dimethylamino-2-chloropropane or 1-chloro-2-dimethylaminopropane must be due to the rearrangement of the chloroamine in the presence of the diphenylacetonitrile anion. The ratio of the amino-nitrile isomers obtained varies with the nature of the basic group of the chloro-amine employed in the alkylation.
8 The following chart has been taken from the article referred to and is reproduced by permission of the publishers of the Journal of the Chemical Society
9 This thermal rearrangement contradicts the work of Attenburrow, et al. ([14] ).
The condensation between diphenylacetonitrile and 1-dimethylamino-2-chloropropane according to the German process gives rise to two isomeric nitriles IIIa and IIIb in approximately equal amounts.
The Grignard reaction between ethylmagnesium bromide and the mixture of isomeric nitriles, followed by the ordinary acid hydrolysis, yields methadone hydrobromide as a precipitate and leaves the ketimine of isomethadone (XIII, as hydrohalide salt) in the aqueous Mother liquor. This ketimine can be converted to isomethadone by prolonged boiling with concentrated hydrochloric or hydrobromic acid.
Diphenylacetonitrile: The mode of preparation of this intermediate is not specified in P B Report 981 ([1] ). However, need for it in the preparation of Methadone and certain analogues has stimulated the development of several very satisfactory syntheses ([15] ).
Condensing agent: In place of sodium amide, lithium amide ([16] ), potassium tert-butoxide ([6] ) and sodium hydroxide ([17] ) have been used.
Separation of the isomeric nitriles IIIa and IIIb: This separation was not effected in the original process. The higher melting isomer IIIa, is less soluble than IIIb in hexane ([6] ) or "aliphatic alcohols, saturated aliphatic hydrocarbons and aliphatic ketones" ([18] ). On the other hand IIIb forms the more insoluble salts with P-toluenesulphonic acid ([19] ) or oxalic acid ([18] ).
IIIa, on treatment with ethylmagnesium bromide, followed by hydrolysis of the Grignard complex, yield methadone in 89 per cent yield ([20] ). An improvement, involving essentially the addition of the Grignard complex to acid rather than the usual reverse procedure, is claimed to lead to improved yields (ca. 90 per cent) ([21] ).
As pharmacological data on the methadones were accumulated, it appeared first that DL-methadone and later that D1-isomethadone would be the more therapeutically important structural isomer (the question of optical isomers is considered below). Efforts were therefore directed toward devising syntheses which would yield one or the other isomer alone, and not, as in the German process, a mixture of nitriles leading to two products. To achieve this it was obviously necessary to avoid the use of a halo-tert-amine and to condense diphenylacetonitrile with a potential threecarbon moiety containing a functional group convertible to a properly situated dimethylamino-group.
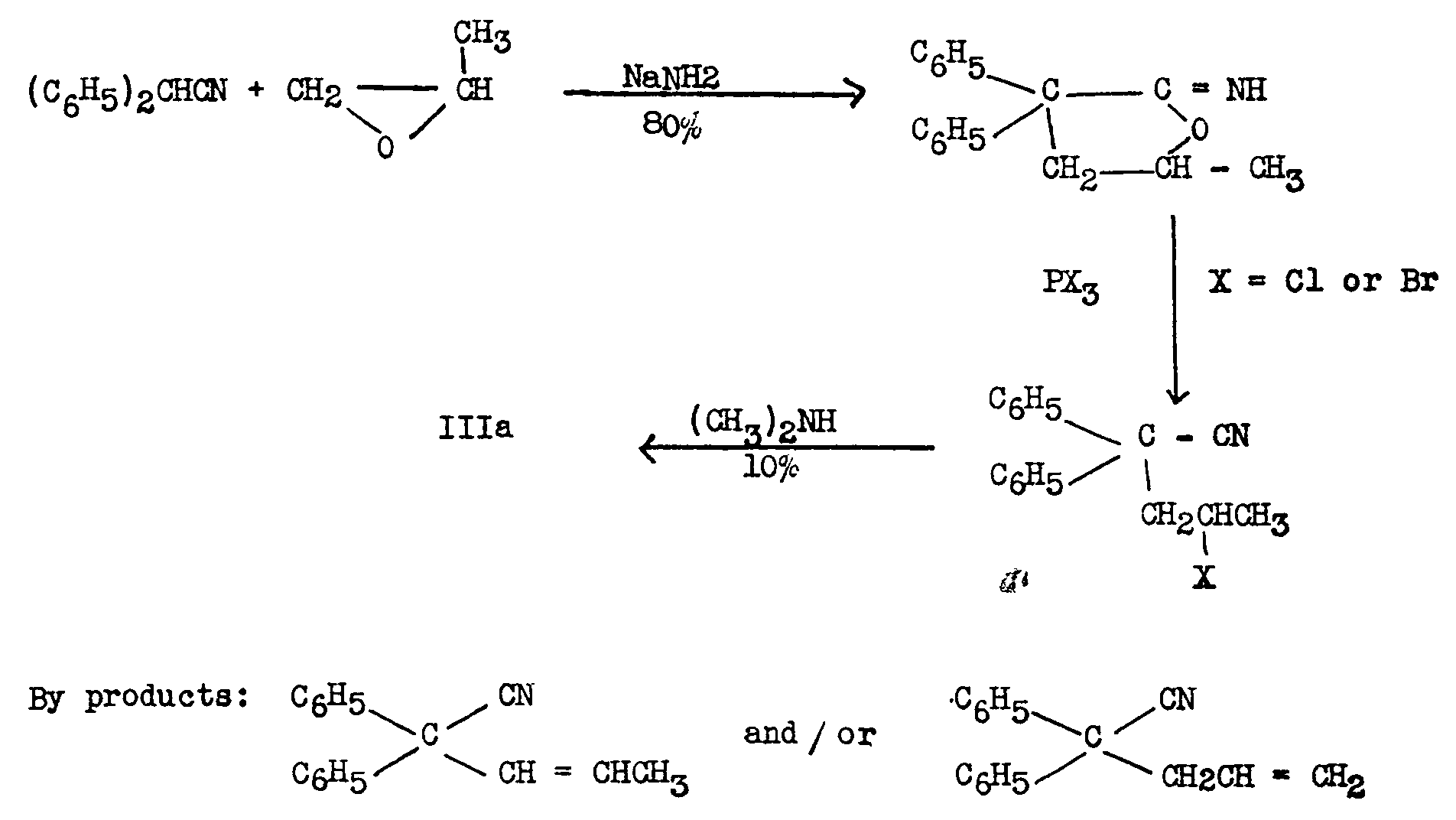
Easton, et al. ([22] ), devised the following sequence leading to IIIa, the nitrile precursor of Methadone. Yields in the last step were poor, an important side reaction being the dehydrohalogenation produced by dimethylamine.
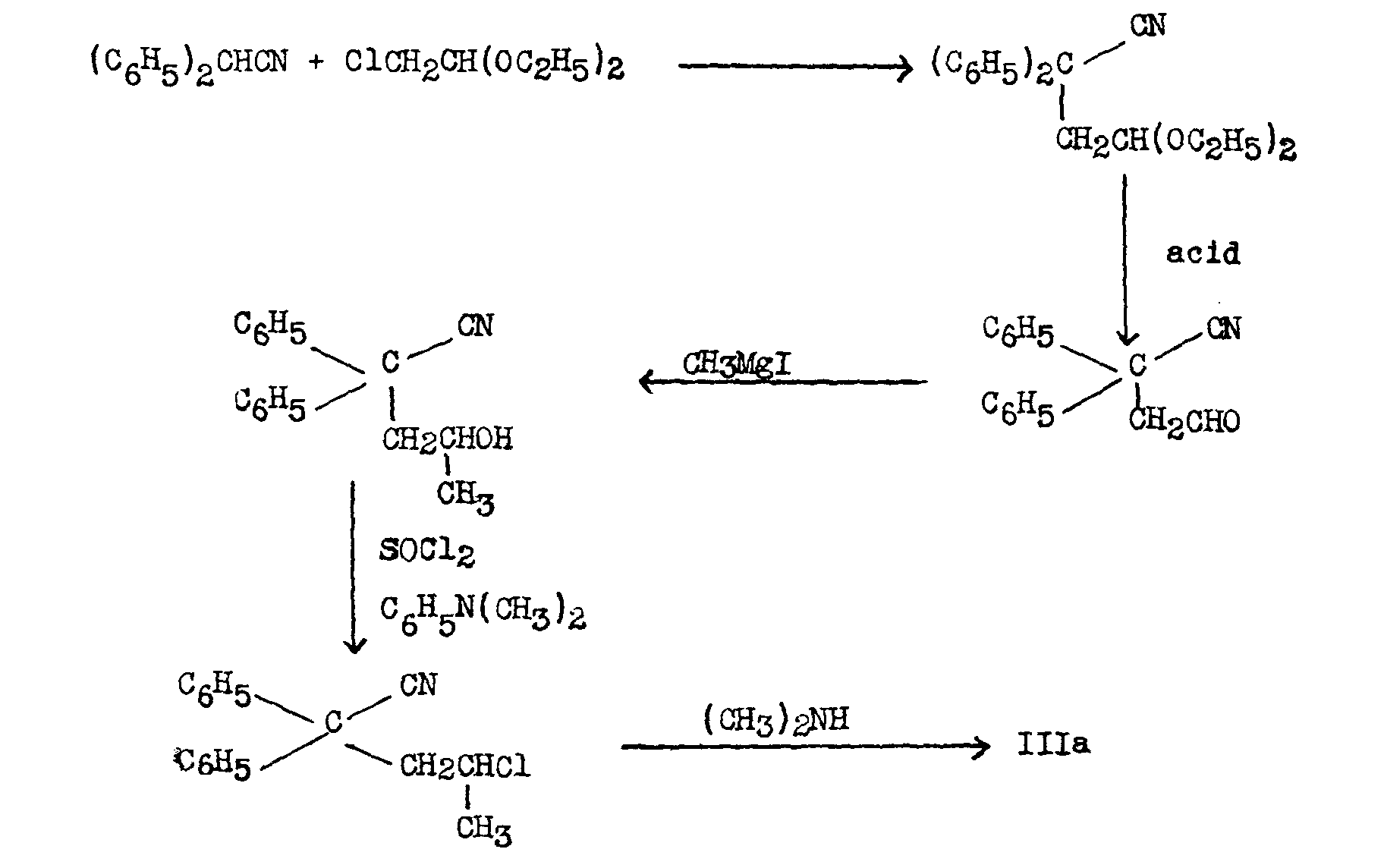
Morrison and Rinderknecht ([23] ) employed the diethylacetal of chloroacetaldehyde as the source of the "sidechain":

Two United States patents have been is-sued recently ([24] , [25] ) on ingenious processes leading exclusively to IIIb, the amino-nitrile precursor of isometha-done, IVb. The steps involved in each are given schematically below:
Attempts to obtain a methadone directly by condensing 4, 4-diphenyl-3-butanone with 1-dimethyamino-2-chloropropane were not successful ([12] ).
Other types of processes have been studied in connexion with the preparation of methadone analogues, particularly the morpholino-analogues XIV.
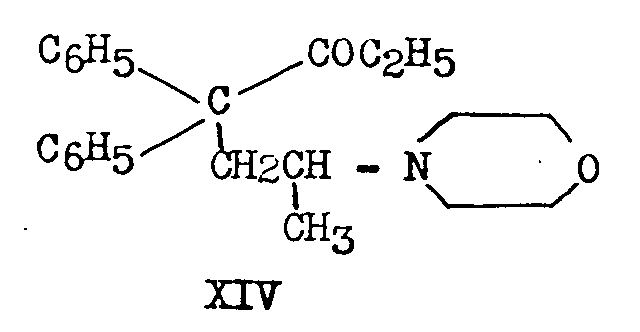
In one process ([26] ) the sodio-derivative of diphenylacetonitrile was condensed with 1-bromo-2-chloropropane. The greater reactivity of bromine relative to chlorine permitted the preparation of 2-chloro-4,4-diphenylvaleronitrate, XV, in 47.5 per cent yield:
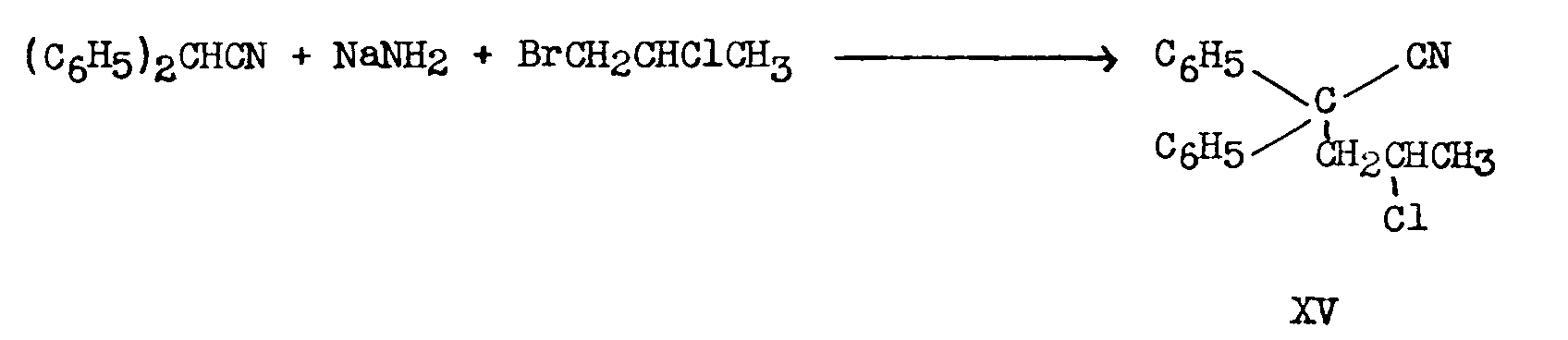
XV heated with morpholine in a sealed tube at 150° for six hours yielded 19 per cent of XVI, which, on treatment with ethylmagnesium bromide produced XIV:
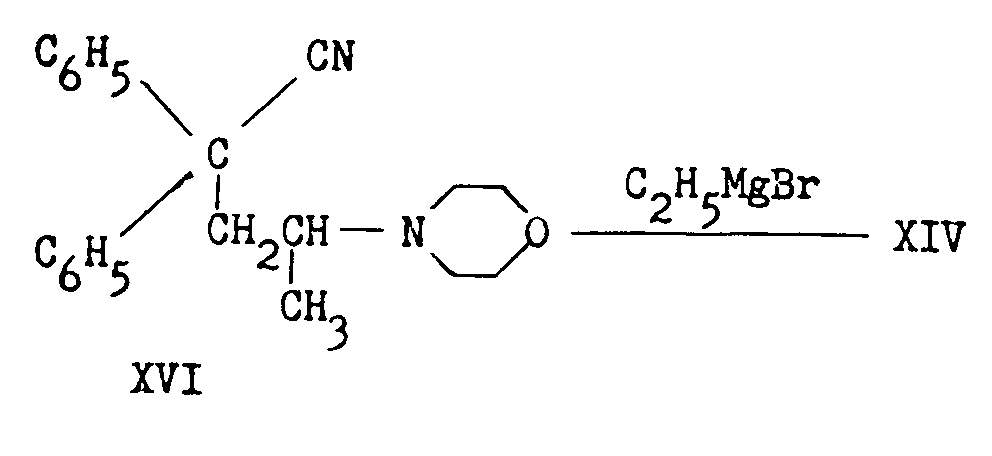
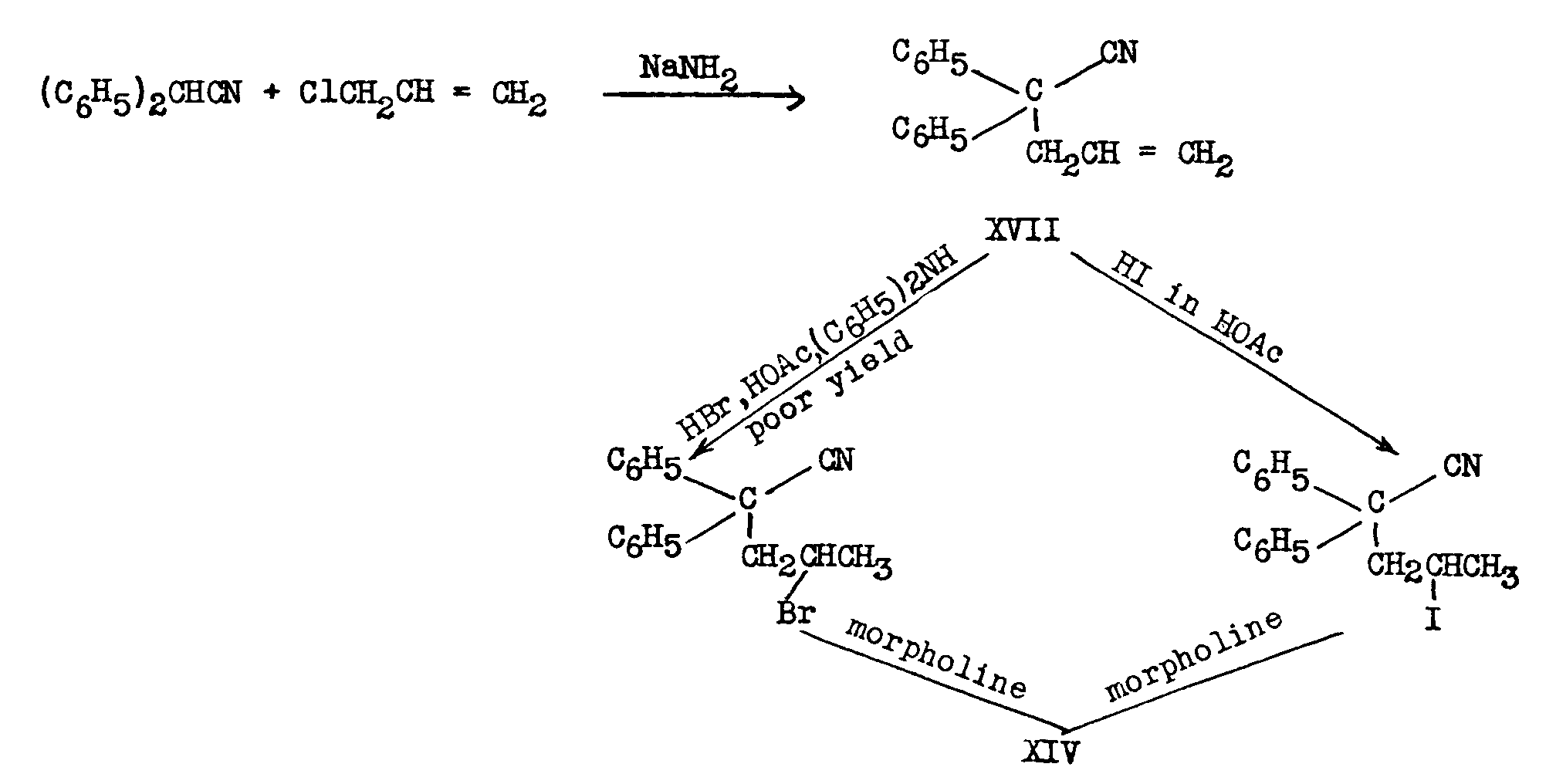
In another process [(27)] diphenylacetonitrile was condensed with allyl chloride. Addition of hydrogen bromide or hydrogen iodide to the resultant olefin (XVII) yielded a 2-halo-4,4-diphenylvaleronitrile which on Grignardization likewise led to XIV:
While DL-methadone may have been resolved in Germany [(3)] ; a description of the procedure used was not available. Brode and Hill [(28)] , who employed D-tartaric acid in acetone, reported the first successfull[10] resolution. By adding pre-formed seeds they precipitated L-methadone-D-acid tartrate; after removing DL-methadone tartrate from the Mother liquor they regenerated the residual bases and obtained pure D-methadone by heating the residue and removing the lower melting eutectic.
Larsen, et al.[(20)] , also reported a resolution of methadone via the tartrates. Howe and Sletzinger (30) treated DL-methadone with D- α-bromo-camphor-π-sulfonate in 80 per cent ethanol and precipitated the salt of D-methadone; from the Mother liquors they prepared L-methadone-D-tartrate.
DL-Isomethadone has been resolved by means of D-tartaric acid [(20)] and by means of p-nitrobenzoyl-L-glumatic acid in isopropyl alcohol [(30)] .
The nitrile precursors (IIIa. and IIIb) of methadone and isomethadone respectively have been resolved by means of D-tartaric acid [(20)] , [(29)] , [(30)] .
It is interesting to note that in the methadone series the L-nitrile on Grignardization yields the L-ketone, while in the isomethadone series the L-nitrile leads to the D-ketone.
The carbonyl group of methadone has only limited reactivity, probably due to steric hindrance. Methadone forms no semicarbazone; it is not reduced by aluminium isoproproxide or the Clemmensen method or sodium amalgam or palladium and hydrogen or Raney nickel at room temperature and atmospheric pressure.[11]
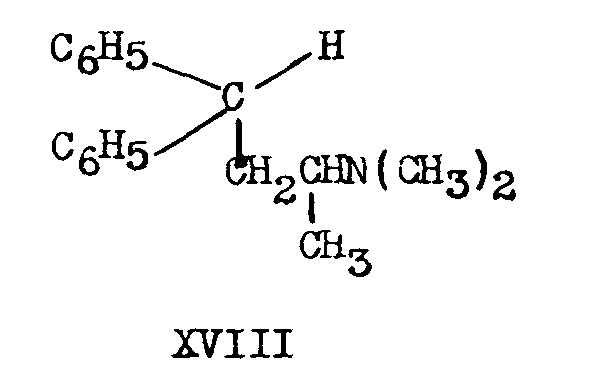
Wolff-Kishner reduction (Huang-Minlon modification) of methadone causes reductive loss of the propionyl group and formation of 2-dimethylamino-4,4-diphenylbutane (XVIII). The latter is also formed when IIIa is heated with potassium hydroxide in triethylene glycol.
10Thorp, Walton and Ofner [(29)] were unable to resolve DL-methadone by means of D-tartaric acid
11 Raney nickel is reported [(33)] to be suitable at high temperature and pressure
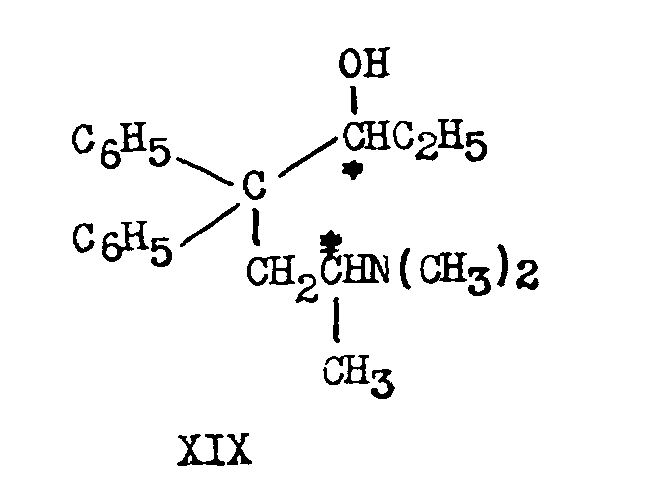
Methadone is reduced catalytically in the presence of platinum or chemically by lithium aluminium hydride to the corresponding alcohol 6-dimethylamino-4,4-diphenyl-3-heptanol, known generally as methadol (XIX).
Since a new asymmetric carbon atom is produced in the course of this reduction, it would be expected that two racemic alcohols would be formed from D1-methadone. This is not the case, however. The reduction, under the conditions mentioned above, is highly stereospecific: DL-methadone leads to a single product, α-DL-methadol, in almost quantitative yield. Reduction of D-methadone yields α-L-methadol[12] . Reduction of L-methadone yields α-D-methadol.
On prolonged catalytic hydrogenation of methadone in the presence of platinum, not only the carbonyl group is reduced to hydroxyl but one of the phenyl groups is reduced to a cyclohexyl group ([16] ).[13]
Reduction of DL-methadone with sodium and N-propanol ([35] , [36] ) is not so stereospecific as the above methods: the product consists of ca. 70 per cent of β-DL-methadol and 10 per cent of α-DL-methadol. By the same method D-methadone yields ca. 40 per cent of β-D-methadol[14] and ca. 5-10 per cent of α-L-methadol; L-methadone yields ca. 40 per cent of β-L-methadol[14] and 10 per cent of α-D-methadol.
A small amount of reductive cleavage accompanies this sodium-propanol reduction to yield 3-dimethylamino-1,1-diphenylbutane.
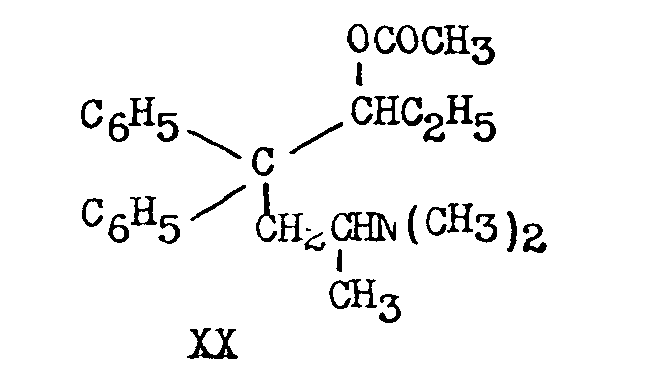
Methadol has been acetylated by acetic anhydride in pyridine (40 per cent yield) or by Houben's method (80-90 per cent yield) or by acetyl chloride in ethyl acetate (90 per cent yield) [(16)] . The product, 3-acetoxy-4, 4-diphenyl-6-dimethylaminoheptane, is often referred to in the literature as acetyl-methadol (XX).
12 Note reversal of direction of rotation.
13 The analogue of methadone in which one of the benzene rings is hydrogenated has also been prepared (34) starting with α-cyclohexyl- α-phenylacetonitrile.
14Note that sign o f rotation is not changed.
When methadol is treated with thionyl chloride it is converted to a mixture of 3-chloro-4,4-diphenyl-6dimethylaminoheptane (XXI) and 4,4-diphenyl-6dimethylamino-2-heptane (XXII) representing respectively direct replacement of hydroxyl by chlorine and cleavage of the elements of water from carbons 2 and 3:
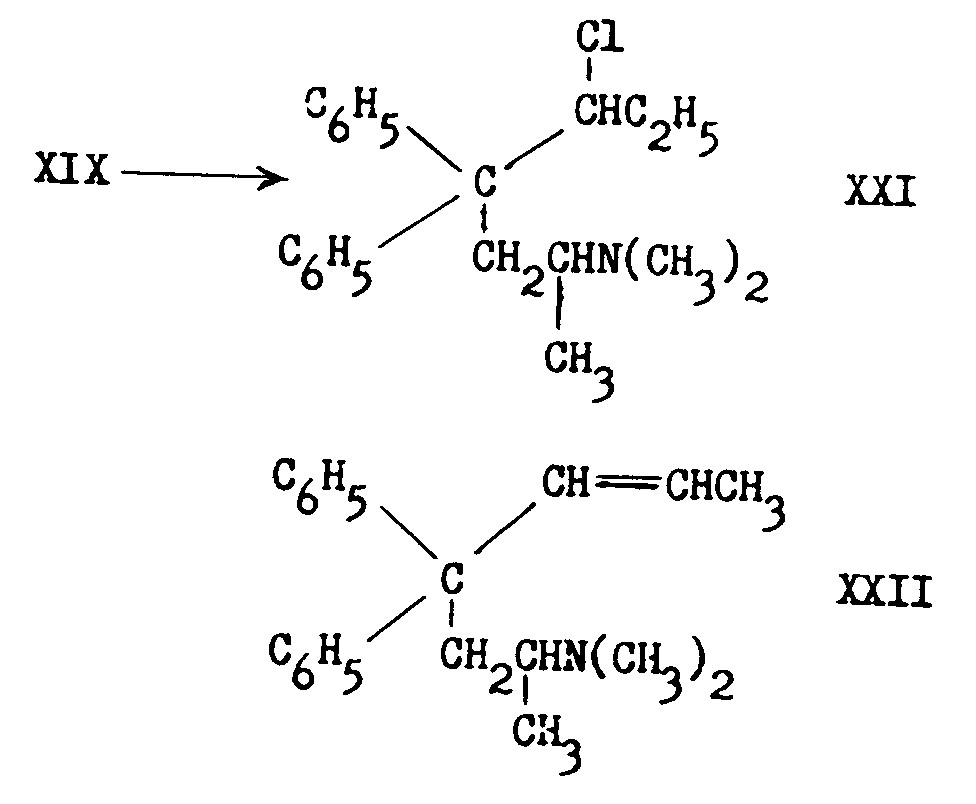
XXII is also formed from XIX by the action of phosphorus pentaoxide in toluene.
The keto group of isomethadone is even more sterically hindered than the keto group of methadone. Isomethadone is not catalytically reduced in the presence of platinum. It is reduced by lithium aluminium hydride to 4,4-diphenyl-5-methyl-6-dimethylannno-3-heptanol, "isomethadol", XXIII.
15The ketimines could be isolated when the phenyl Gnignard or isopropyl Grignard reagent was used ([12] ). Under especially mild conditions the ketimine precursor of methadone was isolated (8b) as acetyl derivative.
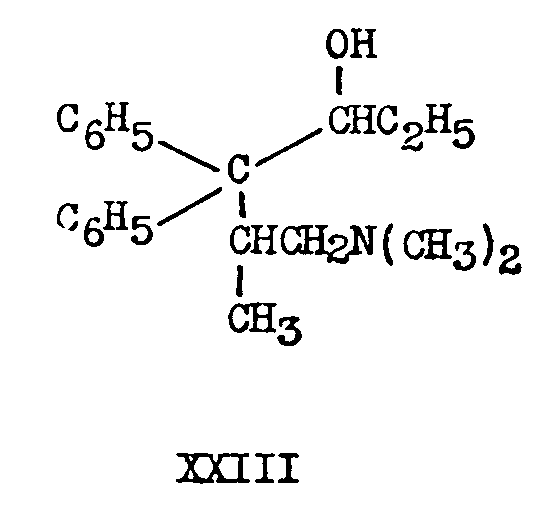
Again the reduction is stereospecific and only onedesignated a- of the two possible racemates is formed from DL-isomethadone. L-isomethadone gave "a-Lisomethadol".
Sodium and propanol reduction of the isomethadones produces a mixture of a-and b-isomethadols [(38)] .
A number of ketones (XXV) have been prepared from the valeronitrile IIIa and the appropriate Grignard reagent. Usually the intermediate ketimine (XXIV) could not be isolated15
The butyronitrile IIIb on reaction with ethylmagnesium bromide yields an unusually stable ketimine (see p. 32-33. This ketimine has been acetylated[ (39)] .
Both IIIa and IIIb are reduced by lithium aluminium hydride (40) to the corresponding aldehydes (XXVIa and XXVIb):
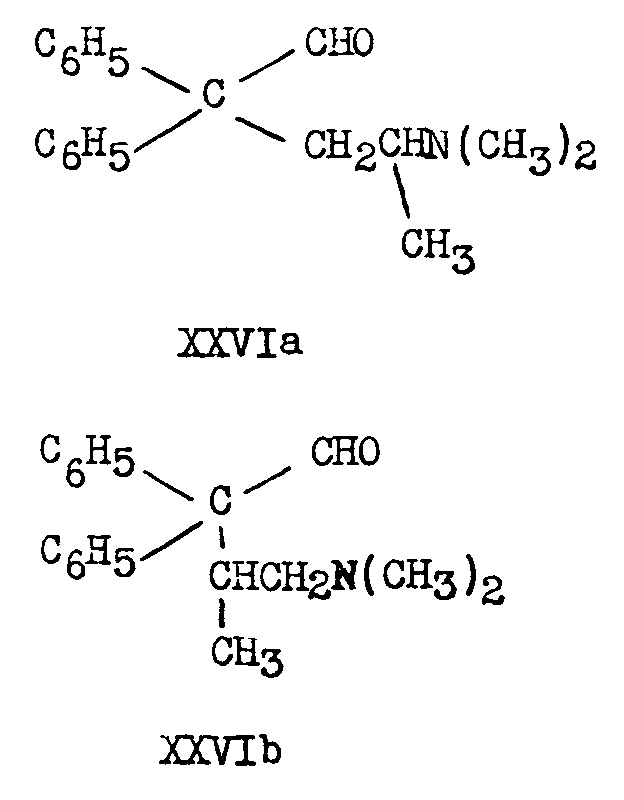
The aldehydes have been catalytically (PtO 2) hydrogenated to the alcohols (XXVIIa and XXVIIb):
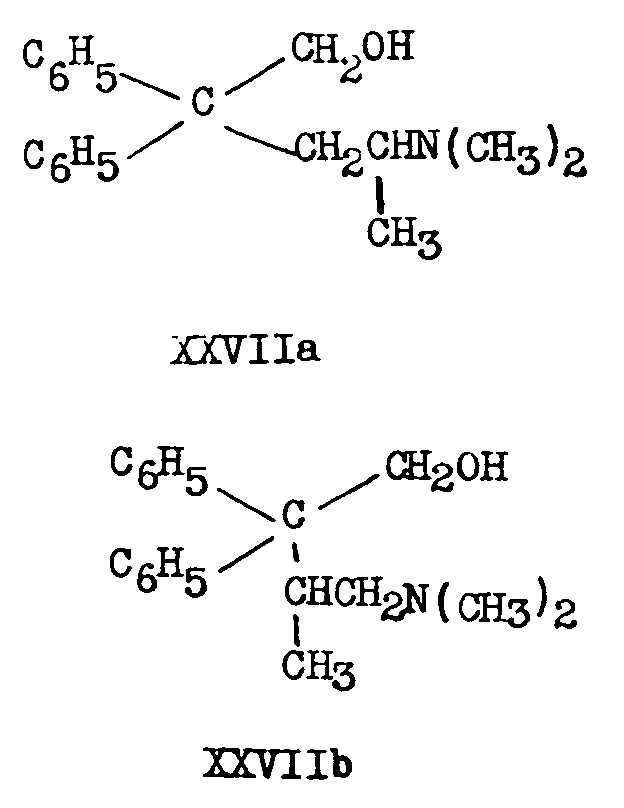
XXVIIa has also been obtained by lithimn aluminium hydride reduction of 2-dimethylamino-4, 4-diphenylvaleric acid (XXVIIIa) or its ethyl ester [(16)] .
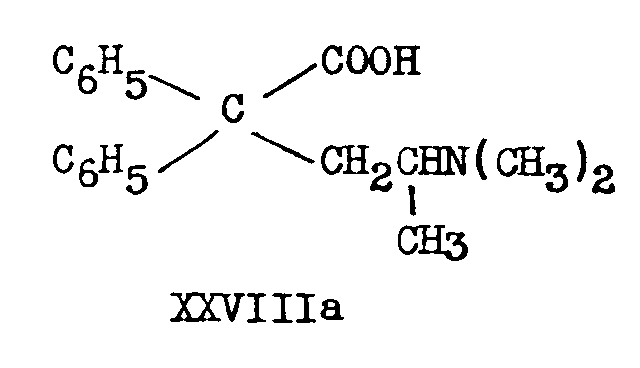
Hydrolysis of IIIa and IIIb with 90 per cent sulphuric acid [(41)] yields the corresponding amides (XXIXa and XXIXb),
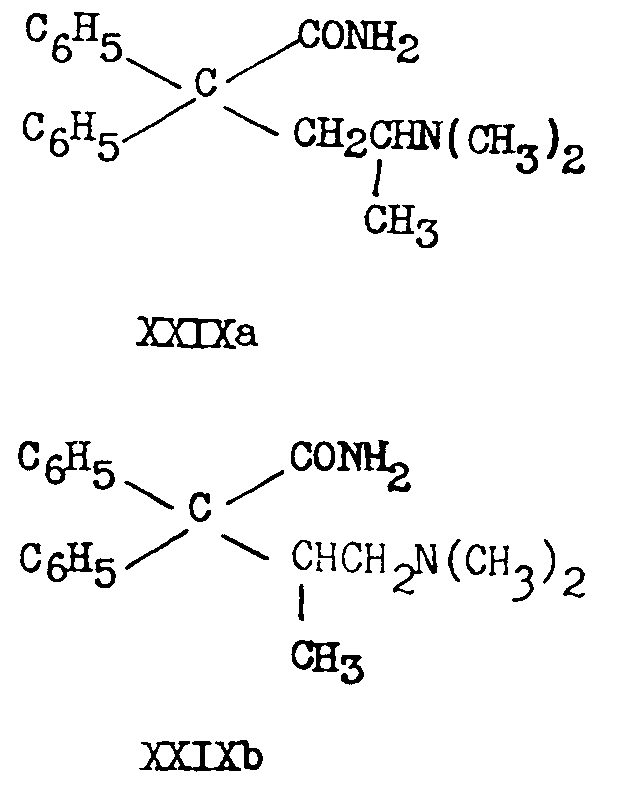
while hydrolysis with 72 per cent sulphuric acid for five hours at 145-150° yields the corresponding acids ([16] ) :16
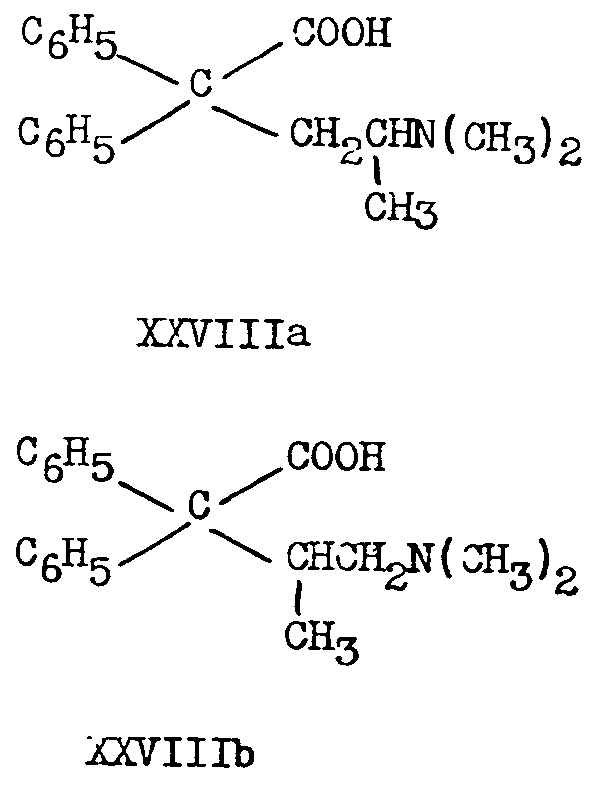
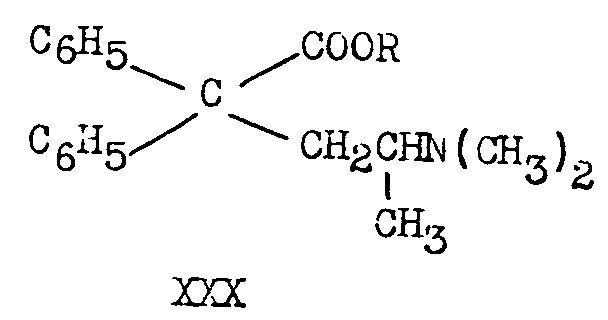
XXVIIIa (as bisulphate) treated with thionyl chloride and the with alcohols R OH (R=Me, Et, Me 2 N CH 2CH 2) yields esters ([12] , [42] ):
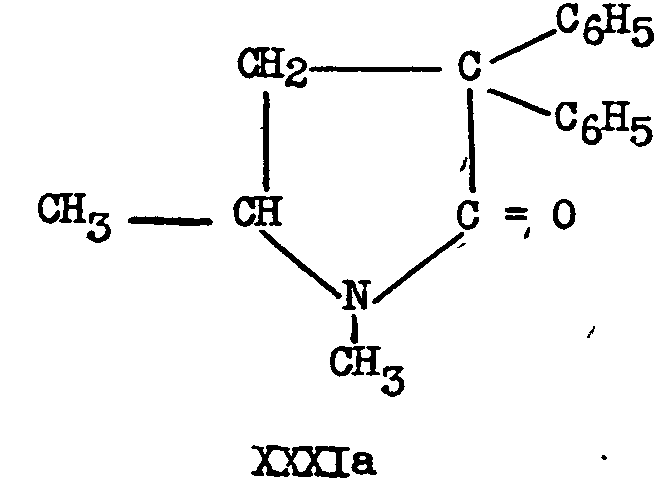
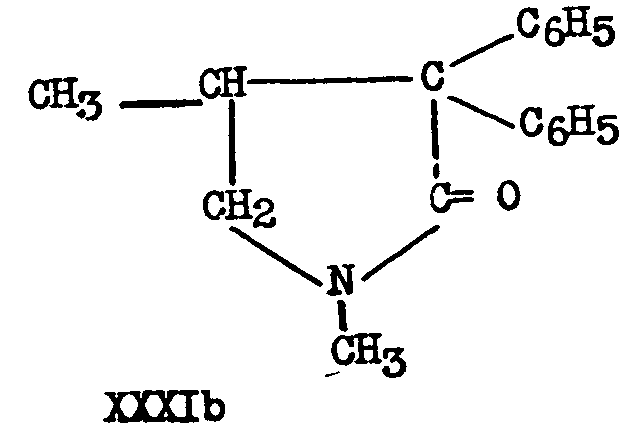
16 Walton, Ofner and Thorp ([12] ) did not succeed in hydrolyzing IIIb to XXVIIIb by sulphuric acid Hydrobromic acid under pressure gave the pyrrolidone XXXIb Cf F F. Blicke and A J Zambito, abstracts of A C.S Meeting, April, 1947, p 3 K.
However, in attempting to form the isopropyl ester of XXVIIIa, Gardner, Easton and,Stevens obtained only 5-dimethyl-3, 3-diphenyl-2-pyrrolidone, XXXIa, the same derivative obtained ([12] ) by the action of thionyl chloride alone on the acid XXVIIIa. Similar ring closures have been reported recently (43) in another connexion.
Attempts (43) to prepare esters of XXVIIIb via the acid chloride led only to the pyrrolidone XXXIb:
Esters of the type represented by XXX on treatment with even an excess of Grignard reagent produce ketones XXV and not tert-carbinols ([35] ).
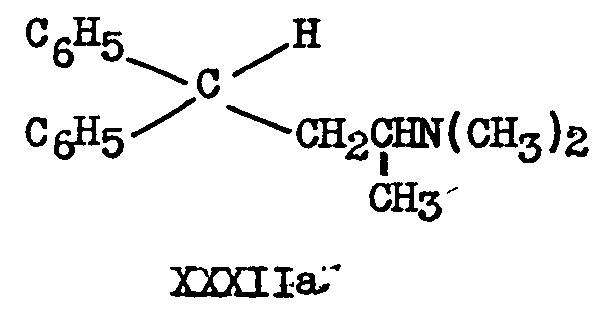
The valeronitrile IIIa on treatment with sodium amide undergoes (reductive) cleavage of the cyanogroup to form 2-dimethylamino-4,4-diphenylbutane XXXlIa.
The same butane derivative is obtained by decarboxylating XXVIIIa at 200° for fifteen minutes ([12] ).
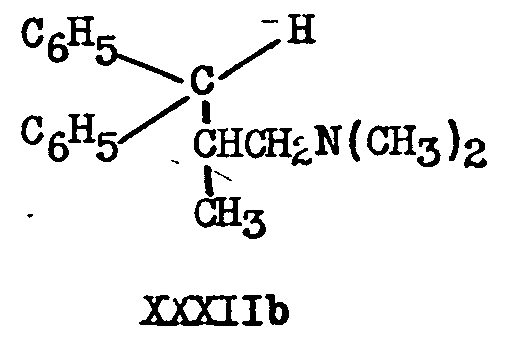
IIIb is decyanated by the action of sodium and isopropyl alcohol (42), but not by sodium amide, to 1-dimethylamino-2-methyl-3,3-diphenyl-propane XXXIIb.
E. C. Kleiderer, J. B. Rice, V. Conquest, J. H. Williams, U.S. Dept of Commerce, Publication Board Report PB-981, July, 1945.
002J. Am. Med. Assn., 138, 651 (1948).
003L. F. Small, Ann. N.Y. Acad. Sci., 51, 12 (1948).
004F. Bergel and A. L. Morrison, Quart Rev, 2, 369 (1948).
005Reference 1, p. 96A.
006E. M. Schultz, C. M. Robb and J. M. Sprague, J. Am Chem. Soc., 69, 188 (1947); J. Am. Chem. Soc., 69, 2454 (1947).
007Private Communication to E. C. Kleiderer cited in reference 3.
008aN. R. Easton, J. H. Gardner and J. R. Stevens, J. Am. Chem Soc., 69, 976 (1947).
008bN. R. Easton, J. H. Gardner, M. L. Evanick and J. R. Stevens, J. Am. Chem. Soc., 70, 76 (1948)
009M. Sletzinger and M. Tishler, U.S. Pat. 2,538,130, January 16, 1951.
010P. Ofner, J. Chem Soc., 1951, 1800.
011E. M Schultz and J. M. Sprague, J. Am. Chem. Soc, 70, 48 (1948).
012E. Walton, P. Ofner and R. H. Thorp, J. Chem. Soc., 1949, 648.
013W. R. Brode and M. W. Hill, J. Am. Chem. Soc, 69, 724 (1947).
014J. Attenburrow, J. Elks, B. A. Hems and K. N. Speyer J. Chem. Soc., 1949, 510.
015C. M. Robb and E. M. Schultz, Org. Syn., 28, 55 (1948); J. Mills, U.S. Patent 2,447,419, August 17, 1948; A. Homeyer and J. S. Splitter, U.S. Patent 2,443,246, June 15, 1948; D. Ginsburg and M. M. Baizer, J. Am. Chem. Soc., 71, 2254 (1949).
016M. E. Speeter, W. M. Byrd, L. C. Cheney and S. B. Binkley, J. Am. Chem. Soc., 71, 57 (1949).
017J. W. Cusic, J. Am. Chem. Soc., 71, 3546 (1949).
018R. W. Stoughton, U.S. Pat. 2,540,636, February 6, 1951.
019K. Pfister, U.S. Pat. 2,497,739, February 14, 1950.
020A. A Larsen, F. F. Tullar, B. Elpern and J. S. Buck , J. Am. Chem. Soc., 70, 4194 (1948).
021W. B Reid and A. W. Schneider, U.S. Pat 2,601,323, June 24, 1952.
022N. R. Easton, J. H. Gardner, and J. R. Stevens, J. Am. Chem Soc, 69, 2941 (1947).
023A. L. Morrison and H. Rinderknecht , J. Chem. Soc., 1950, 1478.
024M. Sletzinger and M. Tishler, U.S. Pat. 2,574,505, November 13, 1951.
025E. M. Chamberlin and M. Tishler, U.S. Patent 2,604,794, August 19, 1952.
026B. A. Hems and J. Elks, U.S. Pat. 2,513,173, June 27, 1950; Brit. Pat. 627,280, August 4, 1949.
027J. Attenburrow, J. Elks, B. A. Hems, K. N. Speyer, J. Chem Soc., 1949, 510
028W. R. Brode and M. W. Hill, J. Org Chem., 13, 191 (1948)
029R. H. Thorp, E. Walton and P. Ofner, Nature, 160, 605 (1947).
030E. E. Howe and M. Sletzinger, J Am Chem. Soc., 71, 2935 (1949).
031A. Pohland, F. J. Marshall and T. P. Carney , J Am. Chem. Soc., 71, 460 (1949).
032E, May and E. Mosettig, J. Org. Chem., 13, 459 (1948).
033R. L. Clark, U.S. Pat. 2,565,592, August 28, 1951
034F. F. Blicke, U.S. Pat. 2,542,466, February 20, 1951.
035M. Bockmühl and G. Ehrhart, Ann., 561, 52 (1948).
036N B. Eddy, E. L. May and E. Mosettig, J. Org. Chem., 17, 321 (1952).
037E. May and E Mosettig, J. Org. Chem, 13, 663 (1948).
038E L May and N. B. Eddy, J. Org. Chem., 17, 1210 (1952).
039L C. Cheney, R R. Smith and S. B. Binkley, J. Am. Chem. Soc, 71, 53. (1949).
040M. Yandik and A. A. Larsen, J. Am. Chem. Soc, 73, 3534 (1951).
041L. C. Cheney, W. B. Wheatley, M. E. Speeter, W. M. Byrd, W. E. Fitzgibbon, W. F. Minor and S. B. Binkley, J. Org. Chem., 17, 770 (1952).
042J. H. Gardner, N. R. Easton and J. R. Stevens, J. Am. Chem. Soc, 70, 2906 (1948).
043P. Lucas, R. L. Clarke and A. Mooradian, U.S. Pat. 2,555,353, June 5, 1951.


