1. INTRODUCTION AND STATEMENT OF THE PROBLEM
2. THE PAPER CHROMATOGRAPHIC SEPARATION OF THE MOST IMPORTANT OPIUM ALKALOIDS
3. THE SENSITIVITY OF PAPER CHROMATOGRAPHIC DETECTION OF OPIUM ALKALOIDS
4. PURITY TESTS OF OPIUM ALKALOIDS BY PAPER CHROMATOGRAPHY
5. CHECKING THE DETERMINATION OF MORPHINE IN OPIUM BY PAPER CHROMATOGRAPHY
Author: J. Büchi, R. Huber, H. Schumacher
Pages: 25 to 45
Creation Date: 1960/01/01
Paper chromatography in its many forms has proved to be a very valuable separation process. Experience has shown that both inorganic and organic ions and molecules of nearly all kinds of chemical substances can be separated even if the substances, as in homologous series, are very closely related chemically. The hydrocarbons are the only exception whose complete insolubility in water precludes separation by paper chromatography. If the sufficiently sharp separation of the substances is achieved, paper chromatograms can be used for isolating known and unknown substances; identifying pure substances and mixtures; purity tests of single substances and mixtures; checking of isolation and determination procedures; quantitative determination of numerous substances; and testing stability of medicinal preparations by the detection and quantitative determination of decomposition products. These general applications show that paper chromatography must be of the greatest value in the investigation of opium alkaloids and opium. Isolation of many of the side alkaloids in opium, which have been described but still insufficiently studied, can be successfully achieved by paper chromatography; moreover, paper chromatography has in fact shown that there must be further alkaloids present in opium which have not yet been described. In testing medicinal preparations this procedure has already been frequently used for identifying types of opium of different origins [ 1] [ 2] , since it is considered possible to determine the country of origin of a sample by establishing the presence of specific side alkaloids, colouring matter and other impurities which are not found in other types of opium. It is also possible to determine the main and side alkaloids and impurities present in a preparation (e.g., opium pulvis, tinctura opii, extractum opii, opium cencentratum, opialum pulvis, ipecacuanhae opiatus or pulvis ipecacuanhae opiatus solubilis, etc.), so that these and other preparations containing opium alkaloids can be identified with certainty [ 3] . Another obvious problem for research was the use of paper chromatography in purity tests of officinal opium alkaloids and salts, which the authors investigated [ 4] with good results. The sensitivity of paper chromatography in detecting opium alkaloids makes it possible to find an impurity of 1% of another alkaloid. This means of detecting small quantities of alkaloids can be used to check determinations of total or individual alkaloids in a drug or preparation. For the determination of morphine in opium, as laid down in Ph. Helv. V,we found in preliminary experiments [ 5] that with this procedure neither a quantitative extraction of morphine from opium, nor the separation of pure morphine is successful; this led us to carry out the experiments described in this paper. The results obtained showed us that all methods of quantitative determination of alkaloid drugs ought to be tested in this way. A quantitative paper chromatographic method for determining morphine in opium is described by Svendsen [ 6] . Paper chromatography is also very useful for testing methods for the preparation of galenicals, because this method makes it possible to observe the progress of extraction of medicinal drugs and the behaviour of the active substances when making up tinctures, fluid extracts, dry extracts and so on. Paper chromatography could be used with advantage in a modern treatment of these problems. It is also very suitable for testing the stability of preparations. It would, for example, be extremely useful if the stability of opium alkaloids could be investigated during the opium extraction stage and storage. On this latter problem a valuable contribution has been made by Panopoulos & Vassiliou [ 1] , who showed that 26 samples of opium from six different countries underwent no change in a year and a half; the chromatograms at the beginning and at the end of the storage period were identical. Similar research should be done on the galenical preparations of opium.
During the last few years we have done experimental work on a few of these multifarious problems. As a basis for further investigations on opium we first studied the separation of opium alkaloids and established the sensitivity of their detection. We then used these data for testing the purity of alkaloid salts and for checking the quantitative determination of morphine and of the most important side alkaloids in various types of opium.
2. 1. Review of the Published Investigations
The opium alkaloids have been the subject of repeated paper chromatographic investigations. Macheboeuf & Munier [ 7] described the separation of morphine, codeine and thebaine by means of a mixture of n-butanol + glacial acetic acid + water (100 + 100 + 50). The alkaloids applied as bases were well separated; but the authors found that under these conditions narcotine and papaverine could not be separated. Codeine and cryptopine behaved similarly. Butanol + glacial acetic acid + water (100 + 30 + saturation) also gave good separation of morphine, codeine and thebaine. By using buffered papers and propanol + water (3 + 1) the authors succeeded in separating thebaine (Rf = 0.75-0.78) from narcotine and papaverine (Rf = 0.94-0.96). Finally they separated morphine, codeine, thebaine, narcotine or papaverine by soaking the paper in a salt solution (M/2 potassium chloride) which contains the same anion as the mobile phase. Using mobile phases capable of unlimited mixing with water such as acetone + water (3 + 1) they secured a separation of morphine, atropine and scopolamine. At the same time they established that the water content of such mobile phases is of significance for the spot form and that over 20% of water is required. A simultaneous separation of morphine and codeine with papaverine and narcotine was not achieved, however, by any of the methods tried. Svendsen [ 6] reported the quantitative determination of morphine from opium and the separation of morphine from other alkaloids. As a mobile phase he used acetic ether + formic acid + water (10 + 1 + 3). The author investigated, with n-butanol + glacial acetic acid + water (20 + 2 + 10), galenical preparations containing opium and tetrapon solutions. He characterized the partly separated alkaloids by means of colour reagents. He was not, however, successful in separating narcotine and papaverine. Mesnard & Boussmart [ 8] tested, with butanol + glacial acetic acid + water (50 + 15 + 15), injections containing, beside morphine, scopolamine, sparteine and strychnine. They were able to identify all of these alkaloids by paper chromatography. Vitte & Boussmart [ 9] examined a syrup, said to contain codeine ane dionine, because the common methods of detection were insufficient. They demonstrated by means of the size of the codeine spot that it was present in a quantity corresponding to that of codeine and dionine. Jatzkevitz [ 10] , Salvesen & Paulsen [ 11] , Wagner [ 12] , Romano [ 13] , Caronna & Bruno [ 14] , Kaiser & Jori [ 15] separated opium alkaloids, partly synthetic morphine derivatives, spasmolytic and other alkaloids, without obtaining a simultaneous separation of papaverine and narcotine. Gore & Adshead [ 16] tested pure alkaloids, and/or their bases and salts by means of n-butanol + formic acid + water (10 + 1 + 10), n-butanol + acetic acid + water (40 + 10 + 50), and n-butanol + propionic acid + water (10 + 1 + 10) as mobile phases, and established that alkaloid bases and salts show an identical migration. Borke & Kirch [ 17] , describe a method for painting glass plates with fluorescent substances. These plates are developed with dioxane. The disappearance of fluorescence shows the position of the alkaloids. The following Rf-values were found: narceine 0.0, morphine 0.38, codeine 0.62, papaverine 0.88, narcotine 0.92. Schute [ 18] separated morphine from the other opium alkaloids with 5% ammonia water as mobile phase. Reichelt [ 19] examined the paper chromatographic behaviour of 12 natural and partly synthetic opium alkaloids by means of formamide as stationary phase, and chloroform, benzene or a mixture of these solvents as mobile phase. With chloroform + benzene (2 + 3) the alkaloids are well separated, but not narcotine and papaverine. Vidic [ 20] investigated the opium alkaloids and spasmolytics from a forensic point of view. He studied the effect of the acid and of water in the system n-butanol + formic acid + water (12 + 1 + 7). He found a drop in the Rf-values during the first few days, and believes that this behaviour can be explained by the degree of saturation of the vapour space in the chromatographic chamber. For the chromatographic separation of opium alkaloids Asahina & Ono [ 21] used n-butanol + ammonia + water (50 + 9 + 15) as mobile phase and found the following Rf-values: narceine 0.58, morphine 0.72, codeine 0.93, narcotine 0.95, and papaverine 0.94. Schultz & Strauss [ 22] attempted to use paper chromatography for an analysis procedure. They investigated numerous alkaloids of the DAB 6 and its supplement (including morphine, codeine, papaverine and narcotine) by means of n-butanol + acetic acid + water (100+10+40), (100+20+53.5), (100+30 + 93.5); n-butanol + formic acid + water (120 + 10 + 70), and n-butanol + 0.1 N hydrochloric acid (50 + 50) as mobile phases. The separation of narcotine and papaverine, however, was achieved with none of the systems proposed. These authors indicate also the calculation of an Rf-value, used for the demixing mobile phase (n-butanol + hydrochloric acid). Mannering [ 23] deals with the paper chromatographic detection of opium alkaloids in urine and animal tissues. Isoamyl alcohol + concentrated ammonia + water (10 + 1 + 5) gives no better separation than the much employed n-butanol + acetic acid + water mixture. Tuderman & Krogerus [ 24] separated various alkaloid salts including morphine hydrochloride, codeine phosphate and papaverine hydrochloride. As mobile phases they used n-amyl alcohol + formic acid + water (100 + 18 + 4) on papers buffered with M/7 potassium phosphate, and as second mobile phase: isopropyl alcohol + water (75 + 25). They found the following Rf-values: morphine hydrochloride 0.16, codeine phosphate 0.33, and papaverine hydrochloride 0.94. Krogerus, Rautiainen & Westerlund [ 25] separated, by means of dioxane + formic acid + water (90 + 0.5 + 9.5), narcotine (Rf 0.89) and papaverine (Rf 0.77), but not codeine (Rf 0.21) and morphine (Rf 0.17). If acetic ether is used as second mobile phase in the same direction, morphine (Rf 0.52) and codeine (Rf 0.63) are separated afterwards. With acetic ether as mobile phase they also separated papaverine (Rf 0.53) and narcotine (Rf 0.85). Curry & Powell [ 26] tested alkaloid fractions in toxicological analyses and, with the system n-butanol + water + citric acid (5 + 5 + 1) on paper buffered with primary sodium citrate, found the following Rf-values: morphine 0.12, codeine 0.16, narcotine 0.47 and papaverine 0.48. Thies & Reuther [ 27] prevent the formation of ester in the solvent mixture butanol + glacial acetic acid + water by the addition of the corresponding ester. This reduces the Rf-values and improves the separability of the alkaloids considerably. With the mobile phase butyl acetate + n-butanol + glacial acetic acid + water (85 + 15 + 30 + saturation) they separated papaverine (Rf 0.57) from narcotine (Rf 0.70). Pfeifer [ 28] gives a method for the separation of the most important poppy alkaloids. First of all on buffered paper, pH 5.5 (citrate buffer) morphine (Rf 0.16), codeine (Rf 0.23), thebaine (Rf 0.61) and narceine (Rf 0.61) are separated from one another and from narcotine (Rf 0.91) and papaverine (Rf 0.92). The latter are finally separated with ammoniacal ether according to the Matthias method [ 29] . Häussermann [ 30] , with an aqueous solution of an electrolyte (250 g of ammonium sulphate, 250 ml of 2 N hydrochloric acid and water to 1,000 ml), separates narcotine and papaverine,
but not morphine and codeine with the following Rf-values: narcotine 0.43 to 0.45, papaverine 0.26 to 0.29, morphine 0.68, codeine 0.67 to 0.69, thebaine 0.47, and narceine about 0.4. The experiments carried out by Bettschart [ 31] for the paper chromatographic separation of opium alkaloids were basic to our investigations. Using water-saturated ether on a paper treated with an M/2 citric acid-phosphate buffer of pH 3.8, he separated narcotine (Rf 0.63) and papaverine (Rf 0.18). But he was not successful with this method in separating the other alkaloids. With a buffer of pH 7.4 he separated, by means of water-saturated ethylacetate, narceine (Rf 0.0), morphine (Rf 0.05), codeine (Rf 0.26), thebaine (Rf 0.82), laudanine (Rf 0.76), landanosine (Rf 0.83) and cryptopine (Rf 0.67). With water-saturated n-butanol he obtained, on a similarly buffered paper of pH 6.8 the following Rf-values: morphine 0.38, codeine 0.56, narceine 0.74, laudanine 0.81, cryptopine 0.81, thebaine 0.84, narco-tine and papaverine 0.96. Experiments with other mobile phases, such as benzene, mixtures of butanol and ether or chloroform, gave less successful separations. Graf & List [ 32] , Mariani [ 33] and Burma [ 34] have recently separated narcotine and papaverine by paper electrophoresis. Horhammer & Leue [ 35] tested tinctura opii simplex DAB6 with n-butanol + acetic acid + water (4 + 1 + 5). Under ultraviolet light they could identify five zones:
|
Rf-value
|
|
|
Blue
|
0.86 |
|
Blue
|
0.74 |
| 0.71 | |
|
Light blue
|
0.57 |
| 0.15 |
Macek, Hacaperkova & Kakac [ 36] examined, in a series of paper chromatographic analyses, the behaviour of about sixty natural alkaloids, including morphine, apomorphine, codeine, oxycodeinone, dihydroxycodeinone, heroin, ethyl-morphine, narcotine, cotamine, thebaine, narceine, crypto-pine and papaverine. They separated the alkaloids using the following mobile phases: chloroform on paper impregnated with formamide + 1% acetic acid, benzene (chloroform) (1 + 1), formamide + benzene (1 + 1), methanol + 5% ammonia + benzene (1 + 1 + 2), n-butanol + acetic acid + water (4 + 1 + 5). Thiess & Reuther [ 37] examined the behaviour of alkaloids with various mobile phases in chroma-tograms. The mobile phases used were butylacetate + n-butanol + glacial acetic acid + water (85 + 15 + 40 + saturation) and butylacetate + glacial acetic acid + water (100 + 60 + saturation). For their experiments they used morphine (0.26, 0.24), codeine (0.39, 0.35), laudanine (0.53, 0.50), papaverine (0.65, 0.56), laudanosine (0.60, 0.53), narcotine (0.74, 0.65) and twenty others. An important contribution towards the systematic analysis of alkaloids with the help of paper chromatography was given by Waldi [ 38] . This author developed a simple process for the identification of alkaloids by soaking four chromatographic papers with a solution of formamide-acetone, drying them, and, after applying the standard alkaloids and the solutions to be tested on the starting line, developing them with four solvents differing in polarity, using the system formamide-cydo- hexane-chloroform-diethylamine. The polarity is varied by altering the ratio chloroform: .cyclohexane. The Rf-values found are converted into R fk Values (values corrected according to the standard) and hR fk values (i.e., hundredfold R fkvalue). Of the opium alkaloids he dealt with narceine, morphine, papaverine, codeine and narcotine. The advantage of this method is that for every alkaloid four values are obtained and it therefore offers means for sure identification. Genest & Farmilo [ 39] described the paper chromatographic isolation, identification and determination of opium alkaloids (morphine, codeine, thebaine and papaverine) in pharmaceutical preparations. The mixtures contained, besides the alkaloid, acetylsalicylic acid, caffein, nicotinic acid, and nicotinic amide or phenacetine. The accuracy of the densitometric determination was good.
As the preceding review shows, the problem of separating the most important opium alkaloids (morphine, codeine, thebaine, narceine, narcotine, papaverine and cryptopine) in one process and on the same chromatogram has remained unsolved.
2.2. Investigations on the Separation of the Opium Alkaloids
By exploiting the details given in published work, and relying on our experiments with other alkaloids, we investigated, in preliminary experiments carried out on tested, pure standard alkaloids, the possibilities of separation. For this we used the following method:
2.2.1. Use of an organic solvent as mobile phase:
Without salt impregnation of the paper,
With salt impregnation of the paper using neutral salts, and
With impregnation of the paper using buffers.
2.2.2. Use of an aqueous solution as mobile phase:
Without addition of a salt to the mobile phase,
Adding a neutral salt to the mobile phase, and
Adding buffers to the mobile phase.
From these preliminary experiments we developed the
Chromatographic procedure A:
Paper. - Whatman paper No. 1, cut in sheets of 10-20/ 48 cm, running direction at right angles to fibre direction; where necessary treated with buffer solutions according to Kolthoff [ 40] .
Mobile phase. - Isobutanol (50 ml), toluene (50 ml) saturated with water. Commercial isobutanol is purified by boiling for two hours with zinc dust and strong alkalis, followed by distillation and redistillation. Toluene is shaken with 7% sulphuric acid for four hours, the sulphuric acid drawn off, the toluene washed with a solution of sodium hydroxide and rectified.
. Application of the alkaloids. - The alkaloids can be used either as bases or as salts, since no difference could be established in the behaviour of the various forms. They are dissolved in chloroform or methanol or in a mixture of these solvents. For slightly soluble alkaloids diluted organic acids or pyridine have been shown to be suitable. Immediately after the preparation the alkaloid solutions are applied by means of a micropipette (Schellbach glass, subdivided in 0.001 ml, calibrated for delivery) on the starting line in quantities of 10 to 100 µg to alkaloid. The starting line is 9 cm from the upper edge of the paper sheet. The solutions are put on a circle of 1 cm diameter. The distance between two such circles should be about 3 cm. Large quantities of alkaloids should be applied in form of a stripe.
Chromatography.- For the descending development we used the Dumas apparatus [ 41] and worked, protected from strong light, in a place free from draughts and as far as possible at constant temperature (19°C to 21°C). On the floor of the chromatographic chamber, which is covered with water to a depth of 1 to 2 cm, stands a wide, flat dish containing the mobile phase. The walls of the chamber are covered with wet filter paper reaching to the floor of the chamber and dipping into the water. The chamber, thus papered, is left for forty-eight hours to allow the saturation of the interior. The paper sheet, prepared as in paragraph 3, is hung in the saturated chamber and conditioned for fourteen hours. Now the mobile phase is poured into the trough so that a depth of 15 mm is reached. The mobile phase, which at once begins to spread in the paper, is allowed to progress for 30 cm (about four hours and a half). Next the damp paper sheet is taken out of the chamber, the line reached is marked and the paper dried at room temperature (15-20 minutes).
Detection of the alkaloids on the paper. - The dried chromatogram is first examined under ultra-violet light and the fluorescing alkaloid spots marked by accurate outlining. Next the chromatograms are sprayed with reagents suitable for ultra-violet or visual detection. If an ultra-violet inspection is not required, the half-dried chromatograms can be sprayed at once with the reagents. This procedure is to be recommended, because it gives colour spots with sharp edges on white paper.
Determination of the Rf-value. - The spots which have appeared are outlined with pencil on the dry chromatograms and measured. T--he distance from the starting line to the centre of the spot is measured.
Detection of opium alkaloids on the chromatograph paper is best carried out with Dragendorff reagent in the composition given by Munier & Macheboeuf [ 42]
|
Solution A
|
|
|
Bismuth subnitrate
|
850 mg |
|
Water
|
40 ml |
|
Glacial acetic acid
|
10 ml |
|
Solution B
|
|
|
Potassium iodide
|
8 g |
|
Water
|
20 ml |
|
Spraying solution
|
|
|
Solution A
|
5 ml |
|
Solution B
|
5 ml |
|
Glacial acetic acid
|
20 ml |
|
Water
|
100 ml |
This reagent turned out to be the most sensitive; moreover, all the opium alkaloids investigated gave orange-red to rosy spots of great permanence.
FIGURE 1
Influence of the reaction of the stable phase on the separation of opium alkaloids
(Chromatographic procedure A)
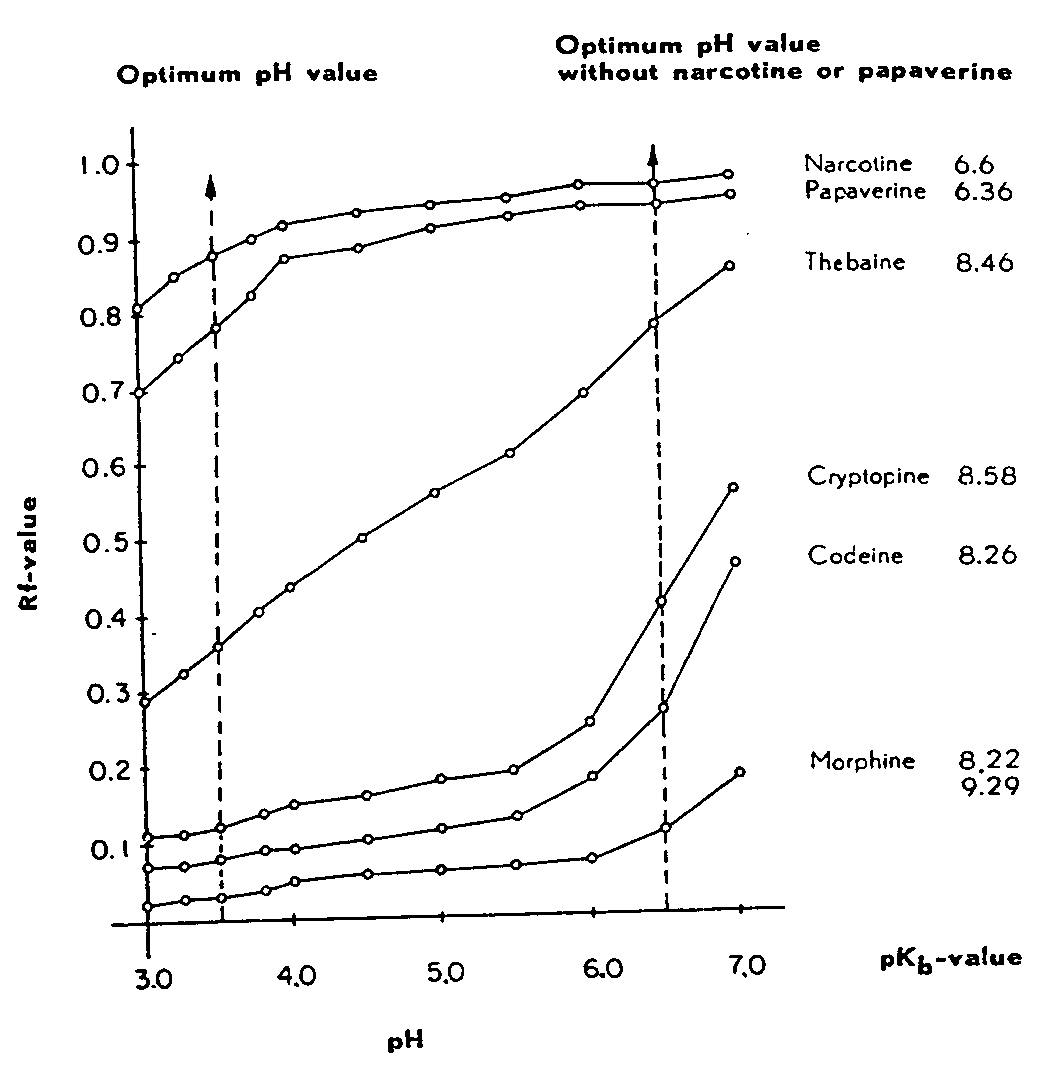
2.3.3 Influence of buffering of chromatographic papers on the separation of opium alkaloids
We used the succinic acid + borax and potassium dihy-drogen phosphate + borax buffer solutions described by Kolthoff [ 40] because of their great stability (absence of carbon-dioxide-sensitive bases), their good buffer capacity, and their small salting-out effect. The chromatographic papers were thus buffered to various pH values between 3.0 and 7.0 and developed according to the chromatographic procedure A. The partition of the opium alkaloids examined is given in figure 1.
All the Rf-values increase as the pH rises. Narcotine and papaverine show a particularly great increase between pH 3.0 and 4.0. Thebaine rises regularly over the entire pH range examined. Morphine, codeine and cryptopine have sharply rising Rf-values towards neutrality. Narceine, which is omitted from this figure for the sake of clarity (pK b-values 2.77 and 9.49) shows up to pH 5.8 an Rf-value above that of thebaine, which is rising regularly with it. At pH above 5.8 (buffer change) the Rf-value drops below that of thebaine (pH 7.0 = Rf-value 0.49). In comparing the Rf-values with the pK-values of the alkaloids investigated no parallelism could be discerned.
The optimum separation of the mixed opium alkaloids was obtained on papers buffered at pH 3.5. Here, morphine, codeine, cryptopine, thebaine, narceine, papaverine and narcotine are distinctly separated. If a solution of narcotine in chloroform is kept for 24 hours or over a longer period a spot is shown on the chromatogram which shines blue in ultra-violet light, is Dragendorff-negative, and has an Rf-value of 0.048. If narcotine or papaverine are not under test, we suggest a separation at pH 6.6. The sequence of alkaloids will then be: morphine, codeine, cryptopine, narceine, thebaine, narcotine or papaverine.
FIGURE 2
Separation of the mixed pure opium alkaloids at pH 3.5
(Chromatographic procedure A)
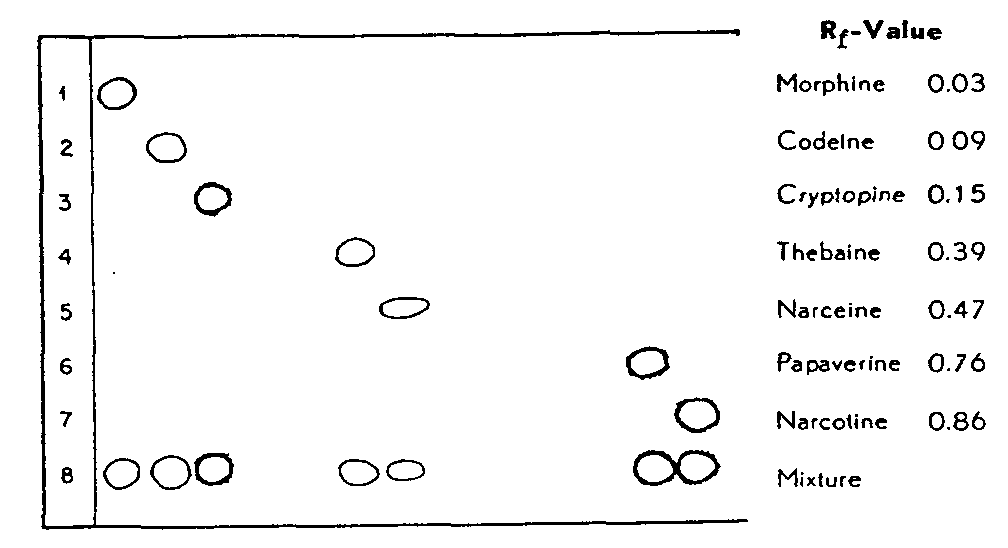
Using chromatographic procedure A, pH 3.5, we were able to separate an Opialum Ph. Helv. V (morphine, codeine, thebaine, narcotine, papaverine and narceine as the hydrochlorides) prepared from our standard alkaloids. A solution containing 500 µg of Opialum was applied drop beside drop on the starting line. In this way we obtained broader but more compact spots, which were clearly visible (fig. 3).
FIGURE 3
Chromatogram of the individual alkaloids and of Opialum Ph. Helv. V at pH 3.5
(Chromatographic procedure A)
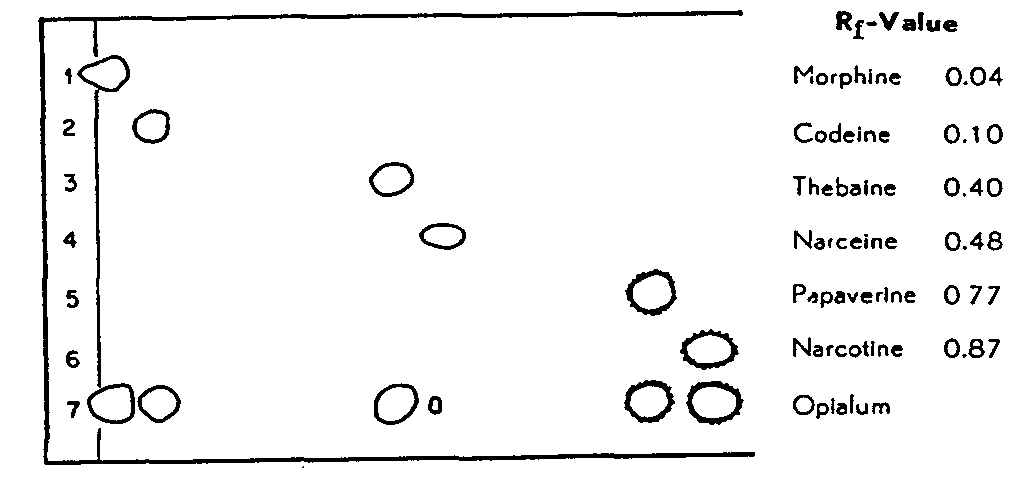
We checked the chromatographic procedure A at pH 3.5 on Extractum Opii, Ph. Helv. V. The separation of the alkaloids was excellent, but we could find no narceine in this preparation. This result was confirmed in the examination of the extract over the entire pH range from 3.0 to 7.0. Pantopon (Roche) gave the same result; narceine is also absent here. In both opium preparations no further alkaloids could be detected.
In table 1 all of the Rf-values are assembled as they were found in investigations on pure alkaloids, on opial and opium extract. They show adequate agreement, which is a further proof that constant Rf-values are obtained by using our method.
These results show that the most important opium alkaloids can be separated with sufficient precision. The Rf-values found differ by more than 0.05 Rf-units and all the alkaloid spots are compact and do not overlap. In the meantime our chromatographic procedure A has been tested by Farmilo, Genest, Clair, Nadeau, Sobolowski & Fiset (43) at pH 3.2.
TABLE 1
Rf-values of opium alkaloids
|
Alkaloid
|
Pure alkaloid
|
Opial
|
Opium extract
|
|
Morphine
|
0.03 | 0.04 | 0.029 |
|
Codeine
|
0.09 | 0.10 | 0.098 |
|
Cryptopine
|
0.15 |
-
|
0.15 |
|
Thebaine
|
0.39 | 0.40 | 0.39 |
|
Narceine
|
0.47 | 0.48 |
-
|
|
Papaverine
|
0.76 | 0.77 | 0.78 |
|
Narcotine
|
0.86 | 0.87 | 0.86 |
These authors obtained approximately the same Rf-values: for all the alkaloids with the exception of narcotine they were very slightly higher, while that for narcotine was 0.03 units lower. Thus the separation of papaverine and narcotine was not so successfully achieved by them as by us. However, in numerous paper chromatographic analyses we have been able to verify our data.
Once we had succeeded in separating the most important opium alkaloids, we determined the amounts of alkaloids which, with the current alkaloid reagents or under ultraviolet light, are just detectable on chromatographic paper. Accurately measured, increasing quantities of alkaloids were applied on papers buffered at pH 3.5, the chromatograms developed and the alkaloids revealed with the reagents listed in table 2. For a comparison with the limit values of detection in the literature, see table 2.
The sensitivity values we established were confirmed by Farmilo, Genest, Clair, Nadeau, Sobolowski & Fiset (46). Since, with the aid of the Dragendorff reagent at least 5 μg of alkaloid can be detected, and results obtained are always reliable, we used this reagent in every case for the detection of very small quantities of alkaloids.
Since preliminary experiments had shown that with the chromatographic procedure A, pH 3.5, very small quantities (5 µg) of alien alkaloid could be separated from relatively large quantities of alkaloid (up to 500 μg), the necessary requirements for a purity test of the opium alkaloids were fulfilled. For this purpose we applied 500 µg of the alkaloid base or salt on the prepared paper sheet, and developed the chromatograms according to procedure A (pH 3,5). The specifications for these purity tests can be formulated as follows:
Morphinum hydrochloricum
" 1.00 ml of basic solution (corresponding to 0.035 g) is diluted with 1.00 ml of alcohol. 0.06 ml of this dilution
Sensitivity of the Paper chromatographic detection of opium alkaloids (corresponding to approximately 525 µg) is applied on the starting line of a paper sheet (Whatman No. 1) and the chromatogram developed according to procedure A. Sprayed with Dragendorff or Kiefer reagent this chromatogram shows only one colour spot, Rf-value approximately 0.03."
|
Alkaloid
|
Sensitivity in μg
|
||||||
|
Dragen-dorff-R
|
Mandel-R
|
Zaffa-roni-R
|
Kiefer-R
|
Ultraviolet detection
|
Author
|
||
|
Morphine
|
5 | 1 | 44 | ||||
| 5 | 20 | 50 | 31 | ||||
| 60 | 24 | ||||||
| 5 | 0.5 | 6 | |||||
| 5 | 20 | 10 | 1 |
*
|
|||
|
Codeine
|
5 | 20 | 31 | ||||
| 30 | 24 | ||||||
| 5 | 15 | 10 | 200 |
*
|
|||
|
Thebaine
|
5 | 20 | 100 | 31 | |||
| 5 | 15 | 10 | 100 |
*
|
|||
|
Narcotine
|
5 | 30 | |||||
| 5 | 20 | 20 | 31 | ||||
| 20 | 45 | ||||||
| 5 | 15 | 10 | 15 |
*
|
|||
|
Narceine
|
5 | 20 |
>50
|
31 | |||
|
50**
|
15 |
>50
|
*
|
||||
|
20***
|
|||||||
|
Papaverine
|
5 | 30 | |||||
| 15 | 24 | ||||||
| 5 | 20 | 0.5 | 31 | ||||
|
<20
|
45 | ||||||
| 5 | 15 | 10 | 3 |
*
|
|||
|
Cryptopine
|
5 | 20 | 31 | ||||
| 5 | 20 | 5 | 50 |
*
|
Dragendorff reagent: Potassium iodobismuthate solution.
Mandel reagent:: Solution of iodine in potassium iodide.
Zaffaroni reagent: Potassium iodoplatinate solution.
Kiefer reagent: Ferrichloride-potassium ferricyanide solution.
Own experiments.
pH below 5.8.
pH above 5.8.
Codeinum
"0.125 g of codeine (accurately weighed) is dissolved in 5.00 ml of alcohol. 0.02 ml of this solution (corresponding to approximately 500 μg) is applied on the starting line of a paper sheet (Whatman No. 1) and the chromatogram developed according to procedure A. Sprayed with Dragendorff reagent this chromatogram shows only one colour spot, Rf-value approximately 0.09."
Codeinum hydrochloricum or phosphoricum
"1.50 ml of basic solution (corresponding to 0.06 g) is diluted with 1.50 ml of alcohol. 0.05 ml of this dilution (corresponding to approximately 500 µg) is applied on the starting line of a paper sheet (Whatman No. 1) and the chromatogram developed according to procedure A. Sprayed with Dragendorff reagent this chromatogram shows only one colour spot, R-f-value approximately 0.09."
Thebaimum hydrochloricum
"1.00 ml of basic solution (corresponding to 0.035 g) is diluted with 1.00 ml of alcohol. 0.06 ml of this dilution (corresponding to approximately 525 µg) is applied on the starting line of a paper sheet (Whatman No. 1) and the chromatogram developed according to procedure A. Sprayed with Dragendorff reagent this chromatogram shows only one colour spot, Rf-value approximately 0.39."
Narceinum hydrochloricum
"0.1 g of narceine hydrochloride (accurately weighed) is dissolved in 10.00 ml of methanol. 0.05 ml of this solution (corresponding to approximately 500 µg) is applied on the starting line of a paper sheet (Whatman No. 1) and the chromatogram developed according to procedure A. Sprayed with Dragendorff reagent this chromatogram shows only one colour spot, Rf-value approximately 0.47."
Narcotinum hydrochloricum
"1.00 ml of basic solution (corresponding to 0.045 g) is diluted with 3.00 ml of alcohol. 0.1 ml of this dilution (corresponding to approximately 500 µg) is applied on the starting line of a paper sheet (Whatman No. 1) and the chromatogram developed according to procedure A. Sprayed with Dragendorff reagent this chromatogram shows only one colour spot, Rf-value approximately 0.86."
Papaverinum hydrochloricum
"1.00 ml of basic solution (corresponding to 0.02 g) is diluted with 1.00 ml of alcohol. 0.1 ml of this dilution (corresponding to approximately 500 µg) isapplied on the starting line of a paper sheet (Whatman No. 1) and the chromatogram developed according to procedure A. Sprayed with Dragendorff reagent or under ultra-violet light this chromatogram shows only one colour spot, Rf-value approximately 0.76."
5.1. Arrangement of the Experiments
After we had elaborated a method for the distinct separation of the most important opium alkaloids and established the sensitivity of their detection, we could check the methods used for the quantitative determination of morphine in opium. The procedure we adopted was to use paper chromatography in examining the different phases in the methods for the determination of morphine:
TABLE 3
Investigated methods of morphine determination in opium
|
Phases of determination method
|
Lime method
Ph. Helv. V
|
Eder & Wäckerlin method (48)
|
Mannich method (50)
|
Fischer & Folbert method (51)
|
Modified Büchi & Huber method
|
|
I. Extraction of morphine from the opium
|
5.00 g of powdered opium + 10 ml of H
2O triturated, then extracted with 2 g of calcium hydroxide + 40 g of H
2O and 26.0 g filtrate obtained
|
1,000 g powdered opium + 50 g H
2O + 2 g calcium hydroxide +manganese sulphate repeatedly macerated
|
1,000 g opium is extracted with 35 g H
2O with addition of lead acetate
|
1,000 g opium is triturated with 5 ml H
2O and the triturate is put on a column of 10 g aluminum oxide (Woelm, acid). Percolation of a total of 35 ml H
2O
|
1,000 g opium + 1 ml H
2O ground and mixed with 5 g aluminum oxide (Woelm, acid), put in chromatographic column containing aluminum oxide (Woelm, acid) and percolated with H
2O until 35 ml of filtrate are obtained
|
|
II. Separation of impurities
|
Addition of 2.5 ml alcohol + 12.5 ml ether + 1 g ammonium chloride
|
Lime extract shaken out with benzene-carbon tetrachloride 1 : 1 in order to remove subsidiary alkaloids. Lime extract buffered with ammonium sulphate
|
|||
|
III. Isolation of morphine
|
Morphine base precipitated and collected quantitatively on a Buchner glass funnel. Morphine dissolved in hot methyl alcohol in order to remove C
aCO
3
|
Morphine shaken out with chloroform - isopropanol 3 : 1 and organic extract put through aluminum oxide (Brockmann); morphine precipitated with NH
4Cl, dissoloved in methanol
|
Precipitation of morphine with chlor-dinitro-benzene in acetone and ammonia. After 12 hours the morphine ether is filtered off
|
Precipitation of morphine with chlor-dinitro-benzene
|
Precipitation with fluoro-dinitro-benzene
|
|
IV. Quantitative determination of morphine
|
Methanolic solution titrated after addition of methyl red with 0.1 N hydrochloric acid
|
Methanolic solution titrated after addition of methyl red with 0.1 N hydrochloric acid
|
Gravimetric determination after 1 hour's drying
|
Gravimetric determination
|
Gravimetric determination or determination in a non-aqueous medium.
|
Extraction of the opium powder (morphine content of the residue of extraction, composition of the opium extract)
Separation of the main quantity of impurities (composition of the morphine solution and of the mother liquor)
Isolation of the morphine (purity of the separated morphine with regard to a side alkaloid content)
Composition of the titrated base (presence of side alkaloids). We used the chromatographic procedure A.
The methods used for the determination of morphine in opium which we investigated are assembled, with an indication of the individual operations, in table 3.
For our investigation we used the following material:
The standard opium alkaloids previously described
The following opium samples
|
Opium I
|
Benares
|
|
Opium II
|
Turkey
|
|
Opium III
|
Iran
|
|
Opium IV
|
Yugoslavia
|
The opium samples I, III and IV originated as samples once sent by the League of Nations to various laboratories for elaborating a method of determination of morphine. They were packed in glass flasks sealed with paraffin-waxed corks and containing 30 g each. These samples were previously used by Eder & Wäckerlin (47, 48) and by Brunner (49) for their investigations. We were able to establish that, compared with earlier determinations (47), the moisture content had increased and the morphine content decreased in these samples.
5.2 Check of the Various Methods
5.2.1. Lime method, Pt. Helv. V
A common feature of the officinal lime methods is the extraction of the opium by a single maceration with lime water and the precipitation of the morphine directly out of the lime with ammonium chloride. An exception is made here by the USP XV, which extracts the opium with pure water. The lime methods aim, in a single treatment of the opium with calcium hydroxide and water, at getting a concentrated opium extract in order to keep the quantity of morphine remaining in the mother liquor as small as possible. Since a part of the liquid used for the extraction is retained by the opium pulp, it is not possible to obtain the entire extract for further treatment, and consequently the morphine can be determined only in an aliquot part of it.
The Ph. Helv. V and other pharmacopoeias allow for the increase in volume of the opium extract due to moisture and the extract substances of the opium by taking 26 ml of filtrate to be the equivalent of 25 ml of original liquor. Since moisture and the extracted matter vary from sample to sample of opium, this correction, as our experiments showed, is only approximate.
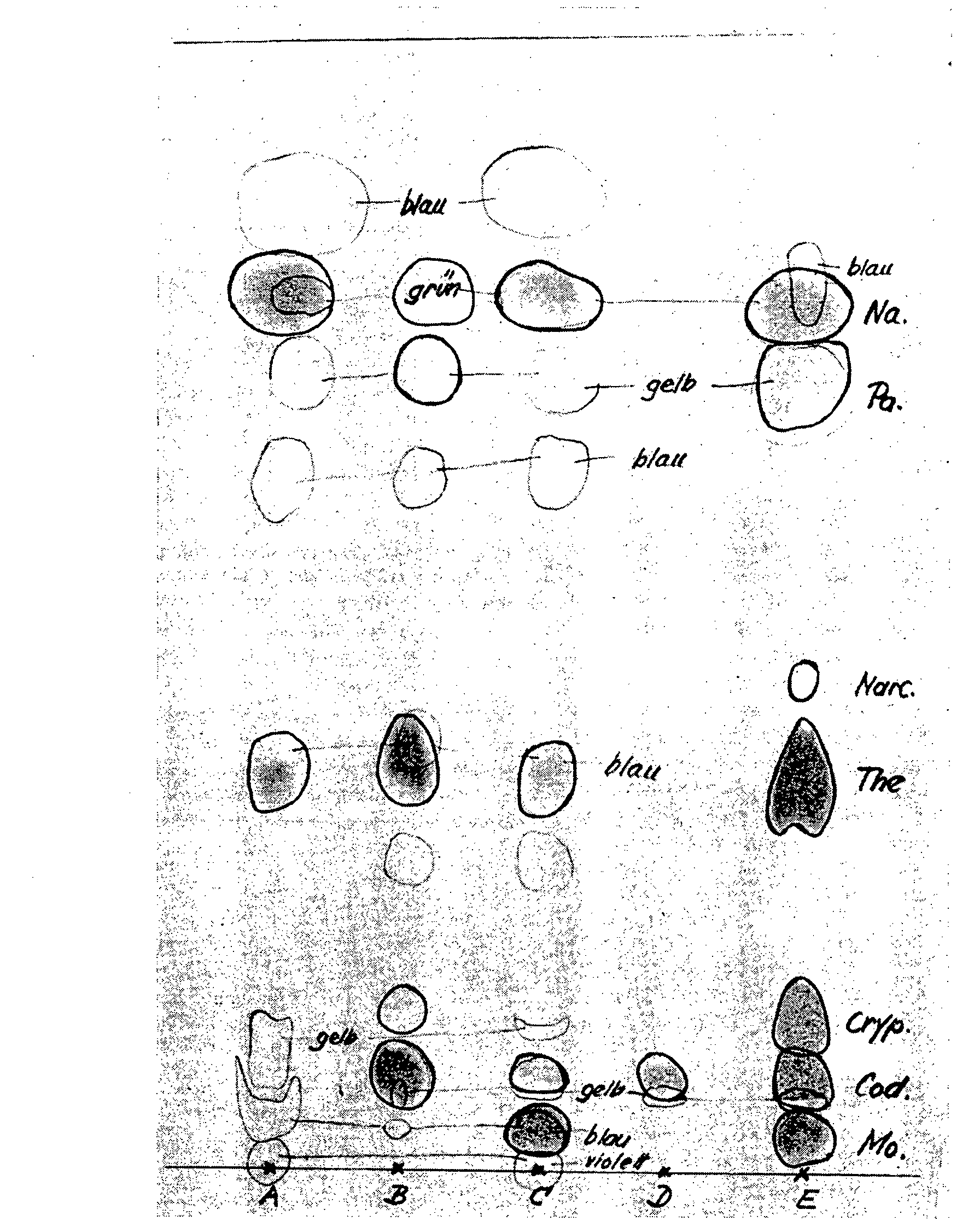
By checking the officinal lime method by paper chromatography (cf. fig. 4) we obtained the following results for the four opium samples:
A. The lime extract (= A) contains not only the morphine, but also considerable quantities of side alkaloids (chiefly codeine, with a little narceine, thebaine, papaverine and narcotine); hence this method of extraction leads to an inadequate separation of the side alkaloids.
B. The ether phase (= B) should dissolve only the side alkaloids, which get into the lime extract. But besides these a small quantity of morphine also gets into the ether and must be regarded as lost.
C. The aqueous mother liquor (= C) should contain no morphine, but considerable quantifies of it remain in this phase and are lost to determination.
D. From the titrated base (= D) the major part of morphine was removed by ion exchange. It was, however, possible to detect considerable quantities of side alkaloids, particularly codeine, and traces of thebaine, papaverine and narcotine.
As a check on the complete extraction of the opium powder by this method, Schumacher (52a) carried out a second mace-ration with lime water and found by paper chromatography no morphine in this macerate; this points to a complete extraction.
Eder & Wäckerlin [ 47] examined the extraction procedure by single maceration of the opium with water and calcium hydroxide. They prepared for this purpose lime extracts of four different opium samples in the proportion 1 : 10, determined their morphine contents according to their shaking out method, and compared the results obtained with those they got by extraction of the opium in the proportion 1 : 50. The average morphine content in the concentrated extract was 10% lower. The authors therefore deny the possibility that a single treatment of the opium with the tenfold quantity of lime water will show the morphine quantitatively. It should, however, be observed that the morphine values obtained according to Ph. Helv. V are not far below those obtained by the Eder & Wäckerlin method [ 47] . On the contrary, in the four opium samples examined they were almost all above them.
The explanation for this may lie in the fact that a less pure morphine is obtained for determination by the method of the Ph. Helv. V. Apart from the correctness of this argumentation, however, there must be a further reason for the difference between the morphine values found by Eder & Wäckerlin [ 47] with their method on the one hand and by the method of Ph. Helv. V on the other. These authors do not say how they prepared the extract 1: 10. Probably they used the same method as for the diluted extract 1 : 50. In this case it may be that a larger quantity of morphine goes into the solution during the half-hour maceration specified by the Ph. Helv. V than in the extraction procedure of Eder & Wäckerlin [ 47] during which the solvent is in contact with the opium for much less time and a complete extraction is attained by repeated use of new quantities of solvent. The repetition and duration of maceration are decisive for this method.
In the case of the Ph. Helv. V specification the question of quantitative extraction is linked up with the question whether 26 ml of filtrate does in fact contain half of the morphine content of the opium. In the case of the Brit. Ph. I958 specification the question is the same with different figures. If the aliquot quantity of 26 ml of filtrate contains in fact half of the morphine present in the opium then the same quantity of morphine should be detectable in the residue and in the rest of the first filtrate. Proceeding from this consideration we extracted various opium samples according to the specification of Ph. Helv. V, took out 26 ml of filtrate and determined, in the manner given below, the quantity of morphine present in it (= flltrate I). The opium residue was then further extracted, the second filtrate combined with the remainder of the first and again the quantity of morphine determined (filtrate II). As a comparison the same opium samples were once again analysed by this method, but instead of carrying this out in two fractions (filtrates t and II), 0.500 g of opium was taken in each case and extracted through repeated maceration with 50 times the volume of lime water (= direct determination). The morphine values so obtained agree very well with the arithmetical mean of morphine contents for filtrates I and II (table 4).
TABLE 4
Morphine content in percentage by different extractions
|
Opium I
|
Opium II
|
Opium III
|
Opium IV
|
|
|
Filtrate I
|
||||
|
(= aliquot part of
|
11.21 | 14.08 | 11.32 | 16.08 |
|
extract 1: 10, single
|
11.08 | 14.12 | 11.22 | 16.22 |
|
maceration)
|
11.26 | 14.20 | 16.15 | |
|
Mean value
|
11.17 | 14.11 | 11.27 | 16.15 |
|
Filtrate II
|
||||
|
(= subsequent extraction
|
11.44 | 14.78 | 11.40 | 16.66 |
|
by repeated maceration
|
11.17 | 14.72 | 11.33 | 16.78 |
|
up to 1: 50)
|
11.59 | 14.60 | 16.54 | |
|
Mean value
|
11.40 | 14.70 | 11.37 | 16.66 |
|
Direct determination
|
||||
|
(= repeated maceration
|
11.27 | 14.36 | 11.58 | 16.45 |
|
1: 50)
|
11.45 | 14.45 | 11.24 | 16.53 |
|
Mean value
|
11.36 | 14.40 | 11.41 | 16.49 |
FIGURE 4
Paper chromatographic check of the opium alkaloids in the different phases of the morphine determination by the lime method, Ph. Heir. V
(Chromatographic procedure A, pH 6.6)
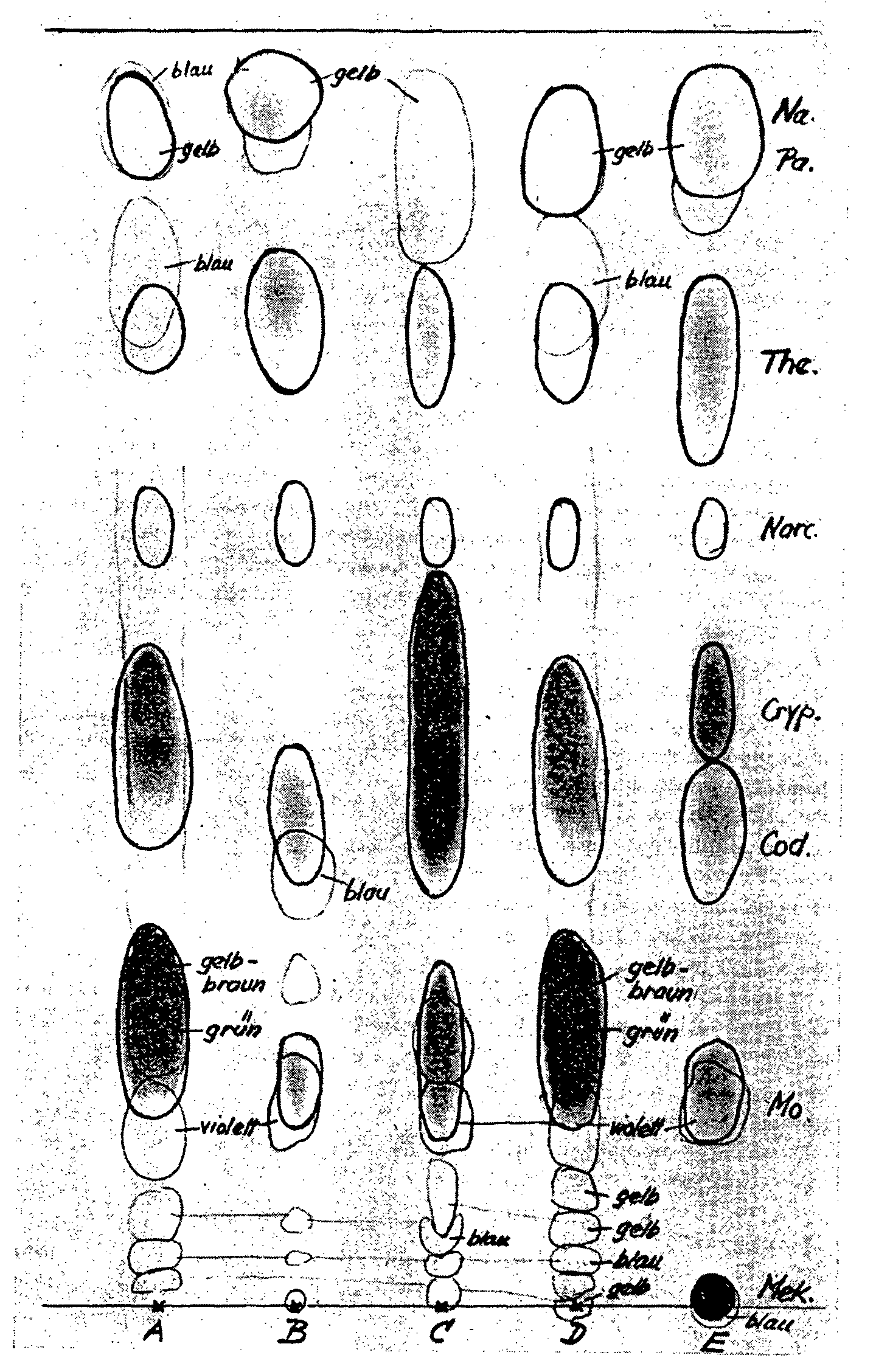
Ph. Helv. V method
(Yugoslav opium, pH 6.6)
Blau = blue
Gelb = yellow
Braun = brown
Griin = green
A = Lime extract
B = Ether phase
C = Aqueous mother liquor
D = Titrated base
E = Standard opium alkaloids
As can be seen from table 4, in determining the morphine in an aliquot part (= filtrate I) of the opium extract, obtained according to the specification of the Ph. Help. V by a single maceration with the tenfold quantity of lime water, a smaller morphine content is obtained than in the extract obtained by repeated maceration with a total of the fiftyfold quantity of lime water (= direct determination). The question remains open whether tiffs difference should be put down to an incomplete extraction or to an improper correction of the increase in volume of the opium extract.
The Ph. Int. I specifies that this increase ill volume should be calculated by determining the moisture and the extract content for each opium sample, but the morphine values so obtained are the same, within the usual limits of error, as those obtained according to Ph. Help. V. From this we deduce that the error due to inexact allowance for the increase in volume is small compared to that due to incomplete extraction. Since these two errors can combine together or balance each other out, this method of extraction is inaccurate.
The loss of morphine in the aqueous mother liquor which has been demonstrated by paper chromatography (figure 4) was subjected to further investigation. Since the Eder & Wickerlin method (48) does not precipitate morphine but shakes it out with chloroform-isopropyl alcohol, this morphine loss does not occur, and a comparative determination according to their method and according to the procedure of the Ph. Helv. V can give information on this point. In three of the four opium samples examined we obtained lower morphine values by the Ph. Helv. V method than according to Eder & Wickerlin (48), whose method counts as one of the most accurate (cf. table 5).
Since in both methods the actual determination of morphine is carried out in the same way by direct titration and since the error made in the extraction procedure of the Ph. Help. V is not big enough to be regarded as responsible alone for the low pharmacopoeia values, the isolation of morphine must furthermore be defective in the way that a larger quantity of morphine remains in the mother liquor than the correction factor of the Ph. Help. V will compensate for. The cause of this may lie in a more or less important hindrance of the precipitation owing to impurities. The correct-hess of this view was proved by determination of the morphine remaining in the mother liquor.
TABLE 5
Morphine content in percentage, according to the methods of the Ph. Heir. Vand of Eder & Wäckerlin [ 48]
(Mean values of several determinations)
|
Method
|
Opium I
|
Opium II
|
Opium III
|
Opium IV
|
|
Ph. Help. V
|
10.18 | 14.36 | 10.70 | 15.58 |
|
Eder R Wiickerlin
|
11.66 | 14.24 | 11.24 | 15.89 |
For this a method had to be chosen which would eliminate the effect of the substances hindering precipitation. It seemed to us that the precipitation of morphine with chlorodinitro-benzene was suitable. Because of the insolubility and high crystallization factor of the morphine-dinitro-phenyl-ether formed, the precipitation of morphine in this form is much less subject to the influence of impurities. These are, moreover, eliminated to a large extent by chromatographic purification and by shaking out. Determinations were carried out as follows:
The mother liquor remaining after filtering off the separated morphine base during morphine determination according to Ph. Helv. V was brought to pH 4-5 by addition of N hydrochloric acid, the ether and alcohol evaporated by moderate warming in vacuo, the mother liquor cooled down, chromatographically purified, shaken out, and the morphine precipitated as dinitrophenylether (method A).
TABLE 6
Morphine content in percentage of the lime extract, Ph. Heir.V
|
Morphine content according to:
|
Opium I
|
Opium II
|
Opium III
|
Opium IV
|
|
Method
Ph. Heir. V
|
||||
|
uncorrected
|
9.04 | 13.22 | 9.56 | 14.44 |
|
Method
Ph. Heir. V
|
||||
|
corrected (+1.14%)
|
10.18 | 14.36 | 10.70 | 15.58 |
|
Method A in the
|
||||
|
mother liquor
|
2.22 | 1.03 | 1.44 | 1.63 |
As can be seen from table 6, the correction factor of the Ph. Helv. V (1.14%), which is meant to allow for the morphine remaining in the mother liquor, is far from agreeing with the morphine quantity present actually in the mother liquor (1.03% - 2.22%). This agrees with the fact that the solubility of morphine is very dependent on the quantity and nature of the impurities present in the mother liquor. Since the quantity of morphine remaining in the mother liquor varies from one opium sample to another, it cannot be covered by a rigid correction factor.
For checking the purity of morphine subjected to determination, the chief method used hitherto was the determination of the methoxyl content [ 47] [ 53] [ 54] , apart from ash analysis and determination of the optical rotation.
With the exception of oxydimorphine and protopine, which, however, occur in opium only in very small quantities, all other side alkaloids of known constitution possess one or more methoxyl groups; morphine which contains side alkaloids also contains methoxyl. The methoxyl content of the morphine, however, gives no detailed information about the qualitative composition of the impurity and allows only rough estimates of the contents of side alkaloids. Thus, for example, one mg of methoxyl corresponds to 9.6 mg of codeine or 4.44 mg of narcotine (1 or 3 methoxyl groups). We carried out purity tests with the help of chromatographic methods. In the case of the Ph. Helv. V method we used as material that part of the filtered off morphine base which could be dissolved with methanol. This solution, in which the morphine is determined by titration, is coloured yellow to brown, and thus contains coloured impurities out of the opium. By paper chromatography [ 4] we were able to detect in it, in addition to morphine, considerable quantities of codeine, and traces of narcotine, thebaine and narceine. For a quantitative determination, we separated the side alkaloids from the morphine by ion exchange chromatography, precipitated them as Reineckates and determined them spectrophotometrically according to Lee Kum-Tatt & C. G. Farmilo [ 55] . For the separation, we used the strongly basic anion exchanger, Dowex 2, which, in the base form, retains morphine from a methanolic solution in contrast to the non-phenolic side alkaloids [ 56] . Separation and determination were carried out as follows:
The titration liquid of the Ph. Helv. V morphine determination was evaporated in vacuo by gentle heating, the residue dissolved in 10 ml of methanol, and this solution put on a column with 10 g Dowex 2 (X-4, 20-50 mesh, base form) in methanol. The column was washed with methanol, until no more codeine could be detected in 5 drops of eluate with 1% sodium tetraphenylboron solution. Speed of flow was 3-5 ml/minute. The eluate was collected in a round-bottomed flask, the solvent distilled off under reduced pressure, the residue dissolved in 5 ml of 2 N acetic acid, and transferred to a small beaker by rinsing with a further portion of 5 ml of 2 N acetic acid. After adding 10 ml of freshly prepared 2% Reinecke salt solution, the mixture was left in the refrigerator for half an hour. The resulting precipitate was collected on a Buchner glass funnel G4, washed with ice water until the filtrate was colourless (using about 20 ml) and freed from the adhering water by passing air through it. The precipitate was then dissolved on the funnel in 2 to 3 ml of acetone, the solution transferred quantitatively to a 10 ml volumetric flask, the funnel rinsed with acetone and the solution diluted with acetone to volume. The absorbance of this solution was measured in an appropriate spectrophotometer at 525 mµ using acetone as blank. The side alkaloid content was calculated as codeine according to the following formula:
|
X =
|
DVM
|
|
ε
|
where X = the amount of alkaloid base found in mg
D = the optical density measured
V = the volume of the solution in ml
M = the molecular weight of the alkaloid base
E = the molar absorptivity of the alkaloid Reineckate (for codeine and morphine Reineckate: ε = 106.5)
In order to check the reliability of the described procedure, several separations of morphine-codeine mixtures were carried out. Codeine was separated and determined as indicated above. Morphine, bound by the anion exchanger, was eluted with 50% acetic acid (speed of flow, 3-5 ml/minute), the eluate concentrated in vacuo to a few ml and morphine precipitated with 1-chloro-2,4-dinitrobenzene (cf. table 7).
TABLE 7
Separation of morphine and codeine with Dowex 2
|
Quantities added
|
Quantities found
|
||
|
Morphine mg
|
Codeine mg
|
Morphine mg
|
Codeine mg
|
| 127.3 | 5.8 | 124.2 | 5.7 |
| 194.0 | 7.3 | 190.4 | 7.4 |
| 300.8 | 18.2 | 297.3 | 18.0 |
TABLE 8
Subsidiary alkaloid content in the titration liquid according to Ph. Helv. V (calculated as codeine)
|
Standard morphine mg
|
Codeine found mg
|
Codeine found %
|
|
|
Opium III
|
239.4 | 16.0 | 6.7 |
| 236.6 | 15.5 | 6.5 | |
|
Opium IV
|
360.5 | 18.4 | 5.1 |
| 363.4 | 18.8 | 5.1 |
In the Ph. Helv. V method the morphine to be titrated contains, as table 8 shows, considerable amounts of side alkaloids. These are titrated as morphine and simulate therefore high values. This is in our opinion a source of serious errors in all the officinal lime methods in addition to the incomplete extraction of morphine from opium and the incomplete precipitation of morphine varying from one opium sample to another.
5.2.2. Eder & Wäckerlin method [ 48]
This method (cf. table 3) is an improvement of the lime method of the Ph. Helv. V, and avoids some of the deficiencies of the latter. The extraction of the opium powder is carried out with a fiftyfold quantity of lime water, and morphine is separated not by precipitation but by shaking out with chloroform-isopropyl alcohol:
In checking this method by paper chromatography we obtained the following results (figure 5):
A. The extracted opium residue was re-extracted with 30 ml of N hydrochloric acid, and this extract (= A) chromatographed. It contained neither morphine nor codeine, but thebaine and papaverine and small quantities of narcotine were present.
FIGURE 5
Paper chromatographic test of opium alkaloids in the various phasesof morphine determination according to Eder k Wäckerlin [ 48]
(Chromatographic procedure A, pH 3.0)
Eder & Wäckerlin method 1940 (Yugoslavia opium pH 3.0)
Blau = blue
Gelb = yellow
Grün = green
A = Second opium extract
B = Benzene-carbon tetrachloride extract
C = Aqueous mother liquor of the morphine precipitate
D = Titrated base
E = Standard opium alkaloids
B. The benzene-carbon tetrachloride extract of the side alkaloids (= B) contained codeine, cryptopine, thebaine, narcotine, and porphyroxine-meconidine, but no morphine.
C. The aqueous mother liquor of the morphine precipitate (= C) contains in addition to side alkaloids a fairly large quantity of morphine, which is lost.
D. From the titrated base (= D) the major part of morphine was removed by ion exchange. In the remaining solution codeine could be detected.
The quantity of morphine remaining in the mother liquor after precipitation has been commonly compensated for by a correction factor obtained in experiments with pure morphine base. But the use of this factor for the determination of morphine in opium is only justified if the purification of the morphine isolated from opium is so complete that it is as pure as the morphine used in establishing the correction factor. That this condition is not fulfilled, and that the correction factor obtained with pure morphine for the determination in opium is too small, results from the fact that the addition of another purifying operation (filtration of the morphine extract with aluminum oxide) in the 1937 method raised the values for morphine by up to 0.5 per cent, although the correction for the losses in the mother liquor remained the same. Our experiments have shown that after the evaporation of the solvent the morphine extract still contains 10%-20% of impurities, and it is therefore to be expected that precipitation will be partly hindered. We determined the quantity of morphine actually retained in the mother liquor in the following way.
The mother liquor obtained by filtering off the precipitated morphine base was transferred directly in a separating funnel and shaken out several times with chloroform isopropyl alcohol 3 : 1. The combined extracts (about 70 ml) were collected in a flask, the solvent distilled off and the residue dissolved in 10 ml of methanol. The separation of the morphine from the side alkaloids was carried out by ion exchange as described (p. 35). The morphine eluted from the column was precipitated as Reineckate and determined spectrophotometrically as described for codeine on page 35.
The results of this determination are given in table 9. While, with a pure morphine base 4.4 mg of morphine remained dissolved in the mother liquor, we found in the mother liquors of morphine determinations in opium (10.4-17.9 mg of morphine) much larger quantities, varying from sample to sample of opium. This result shows once again that the losses in the mother liquor cannot be accounted for by a rigid correction factor. In a further series of experiments we precipitated the morphine not as base but as dinitrophenylether. In this way we found it possible to secure the entire quantity of morphine and to avoid the uncertain factor for losses in the mother liquor. For the detection of any alkaloidal impurities in the morphine subjected to titration, the titrated solution was evaporated in vacuo, the residue dissolved in methanol, and the side alkaloids separated by ion exchange from morphine. By paper chromatography [ 4] we could detect only codeine as an impurity and thus calculate its amount with the aid of the methoxyl content established by Eder & Wäckerlin [ 48] . Compared with the officinal lime methods the codeine content of the morphine to be titrated is relatively low (16% -29%). This underlines the necessity to remove the side alkaloids as completely as possible before the precipitation of morphine as a base.
TABLE 9
Checking of the Eder & Wäckerlin method [ 48]
|
Opium I
|
Opium II
|
Opium III
|
Opium IV
|
|
|
Extract content in %
|
61.30 | 60.59 | 63.87 | 62.13 |
|
pH of mother liquor
|
8.77 | 8.71 | 8.68 | 8.76 |
|
Raw morphine in g
|
0.1260 | 0.1661 | 0.1353 | 0.1870 |
|
Morphine content in %
|
11.7211.60 | 14.1814.30 | 11.2911.18 | 15.9515.84 |
|
Methoxyl content in mg calculated as codeine in mg.
|
0.242.3 | 0.171.6 | 0.302.9 | |
|
Mg of morphine in the mother liquor
|
12.4 | 10.3 | 12.8 | 17.9 |
|
Morphine content in % after precipitation as dinitrophenylether
|
11.40 | 14.36 | 11.38 | 16.32 |
The quantitative precipitation of the pure morphine base offers in all methods so far mentioned the greatest difficulty and is, as our investigations have shown, even in the otherwise well worked out Eder & Wäckerlin [ 48] method, not complete. Moreover, this operation increases considerably the time required for a morphine determination, since 12 to 24 hours must be allowed for precipitation.
It was therefore obvious that one must attempt, in working out new methods of morphine determination, to replace the precipitation of the morphine base as a means of purification by other purifying procedures which are quicker and more sure in effect. A method of morphine determination issued by the Secretariat of the United Nations [ 57] aims to isolate the morphine as pure as possible by a series of extractions, so that it can be titrated without recrystallization.
6,00 g of opium are extracted with the 15-fold quantity of lime water by a single maceration. An aliquot part of the extract is transferred to a separator and after addition of ammonium chloride extracted for 4 to 5 hours with etheracetone 3 : 2. The organic extract carries all the alkaloids.
The solvent is distilled off, the residue dissolved in chloroform and this solution is shaken out with dilute dichloracetic acid, in which morphine is soluble. After addition of ammonia, this solution is shaken out with a mixture of chloroform and isobutyl alcohol (3 : 2) and the solvent distilled off. The residue is dissolved in methanol and morphine determined titrimetrically.
This method is deficient in certain points. The extraction of an unnecessarily large quantity of opium is performed by single maceration, by which, as has been shown for the officinal lime methods, not all of the morphine is extracted. The necessary correction for the increase in volume of the opium extract and the further treatment of an aliquot part of it are further sources of error. This method was subjected to a detailed criticism [ 58] .
Knaffl-Lenz [ 59] attempted, by a special method of extraction and by a series of extractions of the morphine, to isolate it from the opium in so pure a state that it could be directly determined by titration.
3.00 g of opium dried at 105°C are digested with carbon tetrachloride and the solvent filtered off. After addition of sodium sulphate the opium residue is digested several times with water. An aliquot part of the opium extract is shaken out with benzene to separate the side alkaloids, and after addition of sodium bicarbonate with a chloroform-ethanol mixture in order to obtain the morphine. The solvent is distilled off, the morphine remaining is dissolved in an excess of acid and determined by indirect titration.
The method offers no significant advantage in comparison to the extraction methods used hitherto, except that the usual precipitation of the morphine base as a means of purification is omitted or replaced by re-crystallization which can be carried out more quickly. The method of extraction described is very cumbrous, since the opium residue must be repeatedly transferred from the filter to a flask for re-extraction. The treatment of the opium with carbon tetrachloride removes fatty matter, a part of the side alkaloids, and even small quantities of morphine, as we were able to provide in our paper chromatographic tests. The author [ 54] obviously based the development of this method on the insolubility of pure morphine base and of its salts in carbon tetrachloride. This is not, however, the case for the morphine to be found in opium, since the impurities considerably enhance its solubility. The side alkaloids are not completely separated from the morphine by the single shaking out of the opium extract with benzene, so that the morphine to be titrated still contains side alkaloids. The morphine isolated from our opium samples and remaining after the evaporation of the solvent used in the extraction process showed a fairly strong brown colour and therefore contained in addition to side alkaloids some opium impurities. As can be seen by comparison of the values obtained according to various methods shown in table 20, the values obtained by the Knaffl-Lenz [ 59] method show rather low morphine contents. In the following table 10 are given the values found in dried opium by this method and the values calculated for undried opium.
TABLE 10
Morphine content in percentage according to the Knaffl-Lenz method [ 59]
|
Dried opium
|
Un-dried opium
|
|
|
Opium I
|
11.34 | 10.70 |
| 11.27 | 10.63 | |
|
Opium III
|
11.01 | 10.47 |
| 10.92 | 10.40 |
Graf [ 60] attempted by means of chromatography to isolate the morphine from the opium in such a pure state that it could be determined directly by titration.
0.200 g of opium is digested with 1 ml of water, the mixture alkalized, and ground with kieselguhr. The mass is put into a chromatographic tube and percolated first with dichlorethylene to remove the side alkaloids, and then with chloroform/isopropanol 3:1 in order to obtain morphine. The solvent of the morphine fraction is distilled off, the residue dissolved in methanol and the morphine titrated with 0.1 N hydrochloric acid, using methyl red as indicator, to a " strong red colour ".
We tested this procedure, which is very simple in principle and practice, first with a mixture of pure opium alkaloids, which we treated in the same way as is prescribed for opium. Graf [ 60] does not give a detailed definition of the kind of kieselguhr which he used, and only prescribes a type which is as little absorbent as possible. For our experiments we used "Hyflo Super-Cel" (Johns-Manville Co., London). The dichlorethylene used for the elution of the side alkaloids was collected in three fractions of 30 ml each, and each fraction separately examined by paper chromatography [ 4] . Every one of the three fractions contained, in addition to the side alkaloids, some morphine, which in this way is lost to determination. The morphine fraction, on the other hand, was free of side alkaloids. With " Hyflo Super-Cel" as carrier and dichlorethylene as solvent, it is therefore possible to separate by chromatography the side alkaloids quantitatively from morphine, but some morphine is lost. Graf's indication [ 60] to titrate to a " strongly red colour " is imprecise, and is presumably intended to represent a kind of correction factor, which, however, can certainly not be used for every type of opium. In our experiments we titrated the morphine first to the end point of methyl red and then to a strongly red colour. The titration liquid was then evaporated in vacuo, the residue dissolved in 5 ml of 2 N acetic acid, the morphine precipitated as Reineckate and determined spectrophotometrically (cf. p. 35).
As can be seen from table 11, we did not succeed, with the prescribed procedure, to recover the quantity of morphine taken. Since we were able to show by paper chromatography, that morphine was eluted partly with the side alkaloids, the results correspond to our expectations. In opium, the solubility of morphine in dichlorethylene is further increased by the many impurities, so that even higher losses can occur.
TABLE 11
Test of the Graf method [ 60] with pure alkaloids
|
Quantity of morphine base taken mg
|
Quantity of morphine base found mg
|
|||
|
(
a)
|
(
b)
|
(
a)
|
(
b)
|
|
| 28.85 | 25.4 | 25.6 | 23.1 |
Titrated to colour change
|
| 27.0 | 24.5 |
Titrated to " strong red "
|
||
| 25.1 | 23.1 |
Titrated as Reineckate
|
The residue remaining after the distillation of the solvent of opium extracts is coloured dark brown; it is therefore clear that with the morphine some coloured impurities are eluted from the opium, which, if they contain acid or basic groups, will interfere in the titration of morphine. We therefore precipitated the morphine after titration as Reineckate and determined it spectrophotometrically (cf. p. 35). The morphine values found according to this method lie partly below, and partly above, those found by titration (cf. table 12), which shows that one cannot deduce directly the true morphine content from the amount of acid to neutralize the morphine solution. We regard as further sources of error a possible oxydative decomposition of morphine by the addition of ammonia to the hot opium trituration, and the possibility that ammonia is eluted with the chloroform-isopropanol mixture and interferes thus in the titration. In table 12 are given the morphine values obtained according to the Graf method [ 60] . A comparison with the values found by other methods is given in table 20.
5.2.3. Mannich method [ 50]
The method (see table 3) depends on the reaction of the morphine extracted from opium with 1-chloro-2.4-dinitrobenzene to a very insoluble morphine-dinitrophenylether. The extraction of morphine from opium can be done according to Mannich [ 50] in three different ways: by single maceration, by repeated maceration or by percolation. The firstnamed procedure was adopted by the Svenska Farmakopén. Even according to Mannich's own data (50), the extraction of morphine is not complete, since the increase in volume of the opium extract owing to moisture of opium, as well as the extracted substances, are accounted for only by a rigid correction factor. This procedure presents the same deficiencies as the officinal lime methods; incomplete extraction and a not always accurate measurement of the aliquot part of the opium extract. By repeated maceration or by percolation of the opium, on the other hand, all the morphine is extracted.
TABLE 12
Morphine content in percentage according to the Graf method [ 60]
|
Titrated according to Graf
|
Precipitate das Reineckate
|
|
|
Opium I
|
11.12 | 11.2 |
| 11.10 | ||
|
Opium III
|
9.99 | 11.1 |
| 9.80 | ||
|
Opium IV
|
15.73 | 15.9 |
| 15.68 |
The checking of this method by paper chromatography gives the following results (figure 6):
A. The opium residue, extracted by repeated maceration or by percolation with the 35-fold quantity of water, was re-extracted with methanol and N hydrochloric acid (= A). In this second extract neither morphine nor codeine could be detected, but it did contain papaverine and narcotine. The extraction of morphine according to the Mannich method is therefore quantitative.
B. The mother liquor of the morphine ether precipitate (= B) contains no free morphine, from which it can be deduced that morphine is precipitated quantitatively as ether.
C. The morphine-dinitrophenylether (= C) was free from morphine. By digesting a large quantity of morphine-ether with methanol, codeine could be detected.
A considerable quantity of non alkaloidal impurities is precipitated by lead acetate, so that an opium extract obtained by addition of this salt is considerably paler than a pure water extract. Mannich [ 50] states that owing to lead acetate the acidity of the opium extracts is shifted from slightly acid to a nearly neutral point (from pH 3 to pH 6). We were unable to observe such a pH-shift in the opium samples which we examined. After the addition of the prescribed quantity of lead acetate there occurred only a negligible pH-shift towards neutrality; in one case (opium II), on the contrary, we observed a decrease of the pH. As can be seen from the data assembled in table 13, the pH values of the different opium extracts are very near one to another, due to lead acetate, but the weaker bases (narcotine and papaverine) are not precipitated. Accordingly we found, by paper chromatography, in the opium extracts obtained by the Mannich [ 50] method, considerable quantities of these alkaloids.
The precipitation of morphine by conversion to the very insoluble morphine-dinitrophenylether is quantitative, for we were unable to detect by paper chromatography any free morphine in the mother liquor. The morphine-ether formed is, indeed, slightly soluble in the mother liquor. The losses are, however, slight (about 2 mg) and are compensated by the slight impurities in the precipitated morphine ether, so that a correction factor is not necessary. To establish the purity of the precipitated morphine-ether, several authors have chiefly determined the methoxyl content, which permits indeed rough estimates of the side alkaloids content (codeine), but gives no indication of the amount of methoxyl-free impurities. Morphine-dinitrophenylether as a weak base can be titrated with 0.1 N hydrochloric acid [ 50] or in a nonaqueous medium with perchloric acid [ 61] , whereby not titratable impurities can be detected. We checked the purity of the precipitated morphine-dinitrophenylether on the one hand by paper chromatography and on the other by comparison of the titrimetric and gravimetric determination. By titrating with 0.1 N hydrochloric acid and methyl orange as indicator, the end point is only recognizable with difficulty. Methyl red gives a sharper end point, but it occurs too early, so that corrections have to be made [ 50] , [ 54] . We decided therefore on the titration with perchloric acid, where a very sharp end point is obtained with crystal violet as indicator. Only by digesting larger quantities of morphine-dinitrophenylether (about 1 g) with methanol and filtering off the ether, which in contrast to the free alkaloids is very slightly soluble in this solvent, we were able to detect by paper chromatography small amounts of codeine in the concentrated methanolic solution.
TABLE 13
Influence of lead acetate on the pH-value of the opium extracts
|
Ph-value
|
Opium I
|
Opium II
|
Opium III
|
Opium IV
|
|
Aqueous opium extract
|
4.02 | 5.45 | 3.78 | 4.14 |
|
After addition of lead acetate
|
4.65 | 4.86 | 4.58 | 4.73 |
FIGURE 6
Testing of opium alkaloids in various phases of the morphine determination according to Mannich [ 50] by paper chromatography
(Chromatographic procedure A, pH 3.0)

|
A = opium residue re-extracted
|
|
|
Blau = blue
|
B = mother liquor of the morphine-ether precipitate
|
|
Gelb = yellow
|
C = morphine-ether
|
|
D = pure alkaloids
|
As can be seen from table 14, higher morphine values are obtained with the Mannich method than with the other methods mentioned so far, which can be ascribed to the fact that all the morphine present in the opium extract is dealt with. This fact demonstrates the advantage that, compared with the lime methods, no correction factor is necessary. From a comparison of the morphine values obtained by gravimetric and titrimetric determinations of the morphine-dinitrophenylether it can be seen that it is more or less impure according to the opium sample. Vollmer [ 62] pointed out that the nature and quantity of the colloid substances present in the mother liquor account for the fact that the morphine-dinitrophenylether precipitated is capable of carrying down small quantities of side alkaloids. The purification of the opium extract from which the morphine-dinitrophenylether is to be precipitated is, therefore, in this method, the most urgent problem. In order to avoid these sources of errors, van Pinxteren & Smeets [ 54] and Witte [ 63] precipitated the morphine from a lime extract purified with manganese chloride, and Böhme & Strohecker [ 64] purify the opium extract by chromatography on aluminum oxide. This accurate but rather troublesome and lengthy process was quite considerably simplified by Fischer & Folberth [ 51] (see table 3) so that this method seems to be suited for replacing the original method of Mannich [ 50] . The aluminum oxide column retains a large quantity of the ballast substances, recognizable on the brown colouration of the aluminum oxide and the pale yellow colour of the extract. The extraction is, however, rather difficult, since the opium particles settling on the upper surface of the aluminum oxide form a compact mass, through which the water can pass only with difficulty. Thereby, the quantitative extraction of the morphine is prejudiced. It is to this that we ascribe the deviation of the morphine values found with one opium sample (± 0.2%). Neither is the apparative arrangement required for the extraction very advantageous (" the Allihn filter tube containing aluminum oxide is placed on a Witt's filter apparatus which contains an Erlenmeyer flask to collect the filtrate "). Since, however, the method is in principle very simple and can be carried out with a minimum of time, apparatus and reagents, we decided to propose it in modified form as a new pharmacopoeial method.
TABLE 14
Morphine content in percentage according to the Mannich method and titrimetric determination with perchloric acid
|
Single maceration
(Svenska Farmakopén)
|
Repeated maceration
|
|||
|
Gravimetric
|
Titrimetric
|
Gravimetric
|
Titrimetric
|
|
|
Opium I
|
11.56 | 11.21 | 11.91 | 11.52 |
| 11.57 | 11.83 | 11.47 | ||
|
Opium II
|
14.55 | 14.24 | 14.80 | 14.65 |
| 14.65 | 14.92 | 14.78 | ||
|
Opium III
|
11.27 | 11.15 | 11.54 | 11.41 |
| 11.18 | 11.40 | 11.34 | ||
|
Opium IV
|
15.72 | 15.68 | 16.18 | 15.98 |
| 15.85 | 16.23 | 16.02 |
Svendsen & Aarnes [ 65] also use aluminum oxide for the purification of the opium extract, but extract the opium, not with water, but with an organic solvent. The method was adopted by Garratt, Johnson & Lloyd [ 61] and modified in some points. They ground the opium powder, after addition of ammonia and ethanol, with aluminum oxide (standardized according to Brockmann and eluted it in a chromatographic tube with chloroform/isopropanol 3 : 1. The morphine is shaken out of the organic phase with sodium hydroxide solution, precipitated with 1-fluoro-2.4-dinitrobenzene as proposed by Dann & Wippern [ 66] , and determined gravimetrically.
In order to make sure whether 100 ml of the chloroform-isopropanol mixture (Svendsen & Aarnes [ 65] prescribe 240 ml) extract morphine quantitatively from opium, we extracted the opium residue two times with 50 ml of the solvent mixture. In contrast to the first extract these re-extracts were almost colourless, but the first 50 ml contained, as we were able to show by paper chromatography [ 4] , small quantities of morphine, while the second re-extract in the four opium samples examined was free of morphine. In order to determine the morphine content in the first re-extract the solvent was distilled off, the residue dissolved in 30 ml of acetone, and 0.25 g of chloro-dinitrobenzene, 4 ml of 25% ammonia and 36 ml of water added. This solution was still perfectly clear after twelve hours and showed a slight turbidity only after seeding with morphine-dinitrophenylether.
Blanks with 1, 2 and 3 mg of morphine base showed that 2 mg of morphine give a slight turbidity after seeding, while the solution with 1 mg remained perfectly clear, and that with 3 mg showed a slight precipitation. The morphine quantity remaining in the opium residue could therefore have been no more than 2 mg (= 0.2% of the quantity of opium taken). As this finding was the same in all four opium samples, we regard the required quantity of solvent as insufficient to secure a quantitative extraction of morphine. After percolation with 150 ml of chloroform-isopropanol no morphine could be detected by paper chromatography in the residue. In the extracts, emulsions were sometimes formed, which separated often after a long time. The morphine values found by this method correspond to those given by Mannich [ 50] . The greater purity of the precipitated morphine-dinitrophenylether follows from the close agreement between the gravimetric and titrimetric values found. These values are given in table 15.
TABLE 15
Morphine content in percentage according to the method of Garratt, Johnson & Lloyd [ 61]
|
Opium I
|
Opium II
|
Opium III
|
Opium IV
|
|
|
Gravimetrically
|
11.70 | 14.70 | 11.25 | 16.28 |
| 11.64 | 14.59 | 11.30 | 16.37 | |
|
Titrimetrically
|
11.59 | 14.50 | 11.22 | 16.20 |
5.2.4. Establishment of a modified method of morphine determination for pharmacopoeial purposes
The critical examination of the officinal lime methods showed the following considerable deficiencies:
an incomplete extraction of morphine from opium,
the loss of morphine to a degree which cannot be checked during its precipitation,
impurities in the morphine to be titrated consisting of 5 - 10% of side alkaloids,
the use of a comparatively large quantity of opium for the determination, and
too lengthy a process of determination.
Our experiments with the Mannich method [ 50] and its modifications led us to expect that the deficiencies described above could be best avoided by improving this procedure. We therefore revised the different phases of these processes, as described in the following.
Extraction of morphine from opium powder. - (1.000 g of opium powder is ground with 1 ml of water and 5 g of aluminum oxide (Woelm, acid), put on a column of a further 5 g of aluminum oxide (Woelm, acid) and percolated with water, until 35 ml of eluate is obtained. For technical improvements see specification, page 44).
The extraction of morphine from opium proceeds quantitatively in the method given. By paper chromatography we could detect in re-extractions of the column with water and N hydrochloric acid slight quantities of side alkaloids, chiefly narcotine, but no morphine. Filtration through aluminum oxide provides a satisfactory purification of the opium extract, as recognized on its much clearer colour compared with an untreated water extract and by the brown colouration of the aluminum oxide. In table 16 the extract contents of water extracts from various opium samples with and without filtration through aluminum oxide are compared.
TABLE 16
Extract contents in percentage of aqueous extracts with and without filtration through aluminum oxide (Woelm, acid)
(Opium powder = 100 %)
|
Water extract 1 : 35
|
Opium I
|
Opium II
|
Opium III
|
Opium IV
|
|
Untreated
|
52.8 | 47.1 | 64.5 | 58.9 |
|
Filtered through alumi-num oxide
|
48.1 | 35.6 | 49.2 | 42.0 |
Precipitation of morphine with 1-fluoro- or 1-chloro-2.4-dinitrobenzene. - (35 ml of opium extract + 4 ml of 25% ammonia + 0.25 g of 1-fluoro- (or 1-chloro-) 2.4-dinitrobenzene in 30 ml of acetone are shaken and allowed to crystallize).
In order to determine the time required for a quantitative reaction between morphine and fluoro- or chloro-dinitro-benzene we carried out first experiments with a solution of pure morphine hydrochloride, whose exact content on water-free morphine base had been established by the opial method of the Ph. Helv. V. To each 10.0 ml of this morphine hydrochloride solution (= 0.1272 g of morphine base) were added 26 ml of water, a solution of 0.25 g of fluoro- or chloro-dinitrobenzene in 30 ml of acetone and 4 ml of 25% ammonia. The mixture was shaken for a minute and the precipitated morphine-dinitrophenylether filtered off after various time, washed four times with 2 ml of acetone each time and dried for an hour at 80°C. When fluoro-dinitrobenzene was used the morphine-dinitrophenylether was precipitated at once, but with chloro-dinitrobenzene this took place only after some minutes. Many authors prescribe a cooling of the reaction mixture. We therefore carried out our experiments in two series, in which we allowed the morphine-dinitrophenylether to crystallize at room temperature (21°C) and in the refrigerator (+ 1°C) respectively. The results are assembled in table 17.
From the figures given in table 17 it can be seen that the reaction of morphine with fluoro-dinitrobenzene to form morphine-dinitrophenylether is completed after no more than half an hour, while chloro-dinitrobenzene needs at least 12 hours for complete precipitation. Cooling of the mixture holds up the reaction and is, in our opinion, required only after the precipitation is complete in order to lessen the solubility of the morphine-dinitrophenylether. The mother liquors of the incomplete precipitations with chloro-dinitrobenzene were allowed to stand overnight and the morphine-dinitrophenylether which had been precipitated was determined. Thereby the morphine value obtained in the first place could in every case be brought up to 100%.
TABLE 17
Reaction of morphine with 1-fluoro-2.4-dinitrobenzene or 1-chloro-2.4-dinitrobenzene
|
Temperature
|
Time
|
Morphine base found with fluoro-dinitrobenzene
|
Morphine base found with chloro-dinitrobenzene
|
|
%
|
%
|
||
|
21°C
|
?h | 99.8 |
-
|
| ?h | 100.0 |
-
|
|
| 1 h | 100.0 |
ca.60
|
|
| 2 hrs | 100.1 |
ca.90
|
|
| 4 hrs |
-
|
92.4 | |
| 6 hrs |
-
|
98.4 | |
| 12 hrs |
-
|
100.0 | |
|
1°C
|
?h | 99.4 |
-
|
| ?h | 99.8 |
-
|
|
| 1 h | 100.0 |
ca.50
|
|
| 2 hrs |
-
|
ca.80
|
In the precipitation of morphine from opium extracts the possibility of a hindrance of precipitation had always to be reckoned with. In order to show that all the morphine-dinitrophenylether was precipitated after 45 minutes, we filtered it off after periods of 2 and 4 hours. The morphine values found were the same within the limits of error. In no case did a precipitate, insoluble in acetone, form in the mother liquor within the next two days, and paper chromatography also showed no free morphine.
During the precipitation with fluoro-dinitrobenzene the original yellow solution gradually took on a deep orange colour. After about two hours a yellow crystalline precipitate was formed on the bottom, which was clearly distinguishable from the more voluminous morphine-dinitrophenylether. In blanks without morphine the same colour change and the formation of the same crystalline yellow precipitate can be observed. The latter is insoluble in water, slightly soluble in cold alcohol and somewhat more soluble in hot alcohol, but very soluble in acetone. The precipitate, filtered off, washed with water and recrystallized from ethanol melted between 179°C and 180°C (corrected). After the admixture of pure 2.4-dinitro-aniline there was no drop in the melting-point, so that its identity was established. 2.4-dinitro-aniline is produced by the reaction of fluoro-dinitrobenzene and ammonia. With chloro-dinitrobenzene the originally pale yellow solution gradually turned purple, but we did not observe any dinitro-aniline crystallizing out even after several days.
In order to determine any effect of side alkaloids, present in varying amounts in opium, on the reaction between morphine and fluoro-dinitrobenzene, we precipitated the morphine from solutions containing a side alkaloid as well as morphine.
To each 10 ml of a morphine hydrochloride solution, whose exact content on water-free morphine base had been established by the opial method of the Ph. Helv. V, we added a solution of 0.1 g side alkaloid, 26 ml of water, 0.25 g of fluoro-dinitrobenzene in 30 ml of acetone and 4 ml of 25% ammonia, and filtered the precipitate after 45 minutes. In a further series of experiments we determined the morphine in a similar way in a mixture of the pure alkaloid bases with 50.0% of morphine base, corresponding to the opial of the Ph. Helv. V. The results obtained are assembled in table 18.
TABLE 18
Influence of side alkaloids on the reaction of morphine with l-fluoro-2.4-dinitrobenzene
|
Water-free morphine base
|
|||
|
Alkaloid added
|
Quantity taken in g
|
Quantity recovered in g
|
Quantity recovered in %
|
|
0.1 g codeine
|
0.1272 | 0.1270 | 99.9 |
|
0.1 g thebaine
|
0.1272 | 0.1274 | 100.1 |
|
0.1 g papaverine
|
0.1272 | 0.1278 | 100.1 |
|
0.1 g narcotine
|
0.1272 | 0.1273 | 100.0 |
|
0.1 g narceine
|
0.1272 | 0.1269 | 99.8 |
|
Alkaloid mixture
|
|||
| 0.3004 g | 0.1502 | 0.1503 | 100.0 |
| 0.2010 g | 0.1005 | 0.1006 | 100.0 |
| 0.1007 g | 0.0503 | 0.0501 | 99.9 |
From table 18 it can be seen that the side alkaloids present in varying amounts in opium do not influence the reaction of morphine with fluoro-dinitrobenzene; the same applies to chloro-dinitrobenzene which in any case is much less reactive. It must, however, be borne in mind that the content of side alkaloids in the opium extract is only a fraction of the added 0.1 g in the experiments just described. On the other hand the other phenolic bases present in opium may precipitate with morphine. According to Small [ 67] , however, their content in opium is so small (0.005%), that they do not interfere.
Filtration and purification of the morphine-ether precipitate.- (The precipitate is collected quantitatively on a tared Buchner glass funnel G3 by gentle suction, washed with acetone, and dried at 80° C).
According to Mannich [ 50] the precipitate, formed by the reaction of morphine and chloro-dinitrobenzene, is filtered off, washed twice with 2 ml of acetone and twice with 2 ml of water and then dried. This procedure was adopted by all subsequent modifications of the Mannich method. In contrast to chloro-dinitrobenzene, fluoro-dinitrobenzene reacts with the added ammonia under formation of 2.4-dinitro-aniline, which is filtered off with the morphine-dinitrophenylether and therefore makes an intensive washing of the precipitate necessary. Garratt et al. [ 61] require a four-fold washing each with 2 ml of acetone. We checked this procedure by reacting morphine hydrochloride in the usual manner with fluoro-dinitrobenzene, filtering the precipitate off after 45 minutes and washing it in various ways before drying. The determination was carried out gravimetrically and by titration with perchloric acid in glacial acetic acid. The results are set out in the following table 19.
TABLE 19
Washing of morphine-dinitrophenylether formed with fluoro-dinitrobenzene
|
Precipitate washed with
|
Gravimetric determination %
|
Titrimetric determination %
|
Weight of precipitate in g (theor. 0.1673)
|
Morphine-dinitro-phenylether
|
| 0.2684 | 160 | 119.1 | ||
|
1 X 2 ml acetone
|
0.2342 | 140.1 | 109.3 | |
|
2 X 2 ml acetone
|
0.2065 | 123.4 | 101.2 | |
|
2 X 2 ml acetone
|
0.2097 | 122.5 | 101.2 | |
|
2 X 2 ml water
|
||||
|
3 x 2 ml acetone
|
0.1791 | 107.1 | 100.3 | |
|
4 x 2 ml acetone
|
0.1674 | 100.0 | 99.9 |
From all the opium samples we analysed we obtained, after washing with 4 x 2 ml of acetone, a pure, light-yellow coloured morphine-ether, whose determination, carried out in several cases by titration with perchloric acid in glacial acetic acid, always agreed well with the values found gravimetrically.
Since our experiments have shown that the modified procedure extracts all the morphine from opium and converts it quantitatively to the corresponding morphine-ether, and that a very pure substance is obtained suitable for gravimetric or titrimetric determination, we propose the following procedure.
Mannich method, modified by Büchi & Huber. - 1.000 g of opium powder (IV) is ground in a mortar with 1 ml of water to an homogeneous paste and then carefully triturated with 5 g of aluminum oxide (Woelm, acid). In the narrow part of a chromatographic tube (250 mm long, 15-20mm diameter) a pad of cotton wool is put above the tap and the tube half filled with water. 5 g of aluminum oxide (Woelm, acid) is trickled in a thin stream on the water and allowed to settle. Trapped air bubbles are removed by stirring the aluminum oxide with a glass rod. The water on top of the column is allowed to run out until the surnatant water layer is 3 cm above the surface of the aluminum oxide. The opium-aluminum oxide triturate is put in the tube and mixed with the supernatant water by stirring with a glass rod. The last remainders of the opium triturate on the pestle and in the mortar are wiped out with a pad of cotton wool moistened with water, the pad put in the tube and placed on the top of the column, using moderate pressure. The water is now allowed, if necessary under slight pressure, to run out of the chromatographic tube into a glass-stoppered, graduated measuring cylinder (capacity 100 ml) at a rate of one drop per second until the surface of the water reaches the cotton wool pad. Then the filtrate is transferred again into the chromatographic tube, allowed to run, and adding four succesive quantities each of 5 ml of water to obtain a total eluate of 35 ml. 4 ml of 25% ammonia and 0.25 g of 1-fluoro-2,4-dinitrobenzene in 30 ml of acetone are added to the eluate. Then the measuring cylinder is stoppered, shaken for one minute, and allowed to stand for at least 30 minutes at room temperature and for 15 minutes at 5°C. The morphine-dinitrophenylether formed is collected, by gentle suction and rinsing with the filtrate, on a tared Buchner glass funnel G3 and washed with four successive quantities each of 2 ml of acetone. The acetone must be drawn off within at most 15 seconds. If the precipitate has sedimentated too solidly in the glass funnel, it is whirled up with a glass rod in the washing solution.
Gravimetric determination. - The Buchner glass funnel and the precipitate are dried at 80°C for one hour and weighed after cooling in a desiccator. The percentage content of morphine in the opium powder is calculated as follows:
|
Percentage of morphine =
|
g x 63.2
|
|
p
|
where g = weight of precipitate in g
p = weight of opium taken in g
Titrimetric determination. - The washed morphine-dinitrophenylether can also be determined by titration with 0.1 N perchloric acid in a non-aqueous solvent. In this case it is of advantage to use as filter a Buchner glass funnel 1G3 or an Allihn tube 15a G3. The remaining acetone is removed by drawing air through the funnel; the precipitate is dissolved then on the filter in about 20 ml of anhydrous glacial acetic acid, the solution sucked directly into the titration vessel and the filter washed three times with 5 ml of anhydrous glacial acetic acid. 5 ml of acetic anhydride are added and the solution is titrated with 0.1 N perchloric acid potentiometrically or with crystal violet as indicator. Each ml of 0.1 N perchloric acid is equivalent to 0.02852 g C 17H 19O 3N (morphine).
5.2.5. Comparison of the morphine determination methods investigated
In table 20 the morphine contents obtained by the method we propose are compared with those obtained by the other methods. As will be seen, higher morphine values are obtained throughout with our method than with the other methods. We believe that the method we propose extracts on the one hand all the morphine present in the opium and on the other hand determines it quantitatively. The method requires a minimum of reagents and can be easily carried out in half a day.
TABLE 20
Morphine content of the opium samples examined, in percentages as given by the various methods (Mean values of 2-4 determinations)
|
Method
|
Opium I
|
Opium II
|
Opium III
|
Opium IV
|
|
Ph. Helv. V
|
10.18 | 14.36 | 10.70 | 15.58 |
|
Ph. Int. I
|
10.15 | 14.16 | 10.98 | 15.61 |
|
Brit. Ph.1958
|
10.00 | 14.26 | ||
|
U.S.P. XV
|
9.12 | 13.24 | ||
|
Svenska F. XI
|
11.56 | 14.60 | 11.22 | 15.78 |
|
Eder & Wäckerlin (48)
|
11.66 | 14.24 | 11.24 | 15.89 |
|
Knaffl-Lenz (59)
|
10.67 | 10.43 | ||
|
Graf (60)
|
11.11 | 9.90 | 15.70 | |
|
Mannich (50)
|
11.87 | 14.71 | 11.38 | 16.00 |
|
Garratt and collaborators (61)
|
11.67 | 14.64 | 11.28 | 16.01 |
|
Modified method according to
|
12.08 | 14.60 | 11.90 | 16.34 |
|
Büchi & Huber
|
12.12 | 14.74 | 11.84 | 16.29 |
| 12.06 | 14.68 | 11.94 | 16.40 | |
|
12.09
|
14.74
|
11.89
|
16.34
|
G. PANOPOULOS & A. VASSILIOU, United Nations document, ST/SOA/SER.K/16 (1953), 27 (1954), 37 (1955), 42 (1956), 51 (1957).
L.N. MACLEOD, United Nations document, ST/SOA/SER.K/52 (1957).
J. BÜCHI & H. SCHUMACHER, Archiv der Pharmazie, 290, 168, 18l (1957).
J. BÜCHI & H. SCHUMACHER, Pharmaceutica Acta Helvetie, 32, 273 (1957).
J. BÜCHI & H. SCHUMACHER, Archiv der Pharmazie, 290, 191 (1957).
A.B. SVENDSEN, Pharmaceutica Acta Helvetie, 26, 323 (1951). Dansk Tidsskrift for Farmaci, 26, 125 (1952).
R. MUNIER & M. MACHEBŒUF, Bulletin de la Société chimique de France, 852 (1952); Bulletin de la Société de Chimie biologique, 31, 1154 (1949), 32, 209 (1950), 33, 1925 (1951), 34, 213 (1952), 32, 904 (1950).
P. MESNARD & E. BOUSSMART, Bulletin de travaux de la société de pharmacie de Bordeaux, 88, 175 (1950).
G. VITTE & E. BOUSSMART, Bulletin de travaux de la société de pharmacie de Bordeaux, 89, 83 (1951).
H. JATZKEWITZ, Zeitschrift für physiologische Chemie, 292, 94 (1953).
B. SALVESEN & A. PAULSEN, Meddelser fra Norsk Farmaceutisk Selskap, 15, 33 (1953).
G. WAGNER, Pharmazie, 10, 470 (1955).
C. ROMANO, Minerva Medicolegale, 70, 172 (1950).
G. CARONNA & S. BRUNO, Il Farmaco, 10, 497 (1955).
H. KAISER & H. JORI, Archiv der Pharmazie, 59, 224 (1954).
D. GORE & I. ADSHEAD, Journal of Pharmacy and Pharmacology, 4, 803 (1952).
M.L. BORKE & E. R. KIRCH, Journal of the American Pharmaceutical Association (Sc. Ed.), 42, 627 (1953).
J.B. SCHUTE, Pharmaceutische Weekblad, 86, 201 (1951); ref. Deutsche Apotheker-Zeitung, 93, 153 (1953).
J. REICHELT, Pharmazie, 10, 234 (1955).
E. VIDIC, Arzneimittel-Forschung, 5, 291 (1955).
H. ASAHINA & M. ONO, United Nations document, Geneva, June (1955).
O.E. SCHULTZ & D. STRAUSS, Arzneimittel-Forschung, 5, 342 (1955).
G.J. MANNERING, A. C. DIXON, N. V. CAROLL & O.B. COPE, Journal of Laboratory for clinical medicine, 44, 292 (1952).
V.E. KROGERUS & L. TUDERMAN, Eripainos Suomen Apteek-kariyhdistyksen Aikakauslehdest?, 16, 245 (1954).
V.E. KROGERUS, I. RAUTIAINEN & R. WESTERLUND, Suomi Kemis, 28, 117 (1955), Meddelelser Norsk Farmaceutisk Selskap, 17, 198 (1955).
A.S. CURRY & H. POWELL, Nature (London), 173, 1143 (1954).
H. THIES & F. W. REUTHER Naturwissenschaften, 41, 230 (1954).
S. PFEIFER, Sci. Pharm., 24, 84 (1956).
S. MATTHIAS, Naturwissenchaften 41, 17 (1954).
H. HÄUSSERMANN, Archiv der Pharmazie, Berichte der deutschen pharmaceutischen Gesellschaft, 289, 303 (1956).
A. BETTSCHART, Dissertation No. 2408, ETH, Zurich (1954).
E. GRAF & H. P. LIST, Arzneimittel-Forschung, 4, 450 (1954).
A. MARIANI, Pharmaceutische Weekblad, 90, 125 (1955).
D. BURMA, Naturwissenschaften, 41, 19 (1954).
L. HORHAMMER & K.W. LEUE, Archiv der Pharmazie, 288, 377 (1955).
K. MACEK, J. HACAPERKOVA & B. KAKAC, Pharmazie, 11, 533 (1956).
H. THIESS & F.W. REUTHER, Arzneimittel-Forschung, 7, 63 (1957).
D. WALDI, Archiv der Pharmazie, 292, 206 (1959).
K. GENEST & CH. FARMILO, Journal of the American Pharmaceutical Association, 48, 287 (1959).
J.M. KOLTHOFF, Der Gebrauch der Farbindikatoren, 3rd edn., published by Springer & Co., Berlin, p. 148 (1925).
Manufacturer: Arnnold Dumas, Zurich 7/44, Schmelzberstr. 26.
R. MUNIER & M. MACHEBŒUF, Bulletin de la Société de Chimie biologique, 32, 192, 304 (1950).
C.G. FARMILO, K. GENEST, E.G. CLAIR, G. NADEAU, G. SOBOLOWSKI & L. FISET, United Nations document ST/SOA/SER.K.58, 14 (1957).
H. KAISER & H. JORI, Archiv der Pharmazie, 287 (1954).
R. MUNIER & M. MACHEBŒUF, Bulletin de la Société Chimie biologique, 31, 1146 (1949).
C.G. FARMILO, K. GENEST, E.G. CLAIR, G. NADEAU, G. SOBOLOWSKI & L. FISET, United Nations document ST/ SOA/SER.K/ 58, 9 (1957).
R. EDER & E. WÄCKERLIN, Journal of Pharmacy and Pharmacology, 10, 680 (1957).
E. EDER & E. WÄCKERLIN, Pharmaceutica Acta Helvetiae, 15, 227 (1940).
H. BRUNNER., Dissertation, ETH, Zurich (1938).
C. MANNICH, Archiv der Pharmazie, 280, 386 (1942).
R. FISCHER & K. FOLBERTH, Arzneimittel-Forschung, 5, 66 (1955).
H. SCHUMACHER, Dissertation No. 2690, ETH, Zurich (1958).
52a. H. SCHUMACHER, personal communication.
O. F. GRÄNICHER, Dissertation ETH, Zurich (1936).
J.A.C. VAN PINXTEREN & M. A. G. SMEETS, Pharm. Wbl., 85, 48 (1950).
LEE KUM-TATT & C. G. FARMILO, United Nations document ST/SOA/SER.K/49 (1957).
A. FISCHER, Deutsche Apotheker-Zeitung, 97, 23 (1957).
United Nations document, ST/SOA/SER.K/1 (1951).
Do. ST/SOA/SER.K/44 (1956).
E. KNAFFL-LENZ, United Nations document ST/SOA/SER.K/3 (1951).
E. GRAF, Deutsche Apotheker-Zeitung, 91, 797 (1951).
D.C. GARRATT, C. A. JOHNSON & C.L. LLOYD, Journal of Pharmacy and Pharmacology, 7, 914 (1957).
K. VOLLMER, Deutsche Apotheker-Zeitung, 95, 258 (1955).
A.H. WITTE, United Nations document ST/SOA/SEK.K/67 (1958).
H. BÖHME & R. STROHECKER, Arnzneimittel-Forschung, 3, 468 (1953).
A.B. SVENDSEN & E.P. AARNES, Scientia pharmaceutica, 23, 18 (1955).
O. DAHAN & F. WIPPERN, Deutsche Apotheker-Zeitung, 91, 905 (1951).
L.F. SMALL, Chemistry of the Opium Alcaloids, Washington, 1932.


