Introduction
Materials and methods2
Results and discussion
Author: H. N. ELSOHLY, C. E. TURNER
Pages: 51 to 56
Creation Date: 1982/01/01
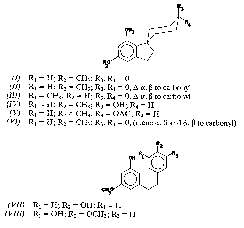
The first reports of the presence of spiro-indans in Cannabis 1were published almost simultaneously by two independent groups [ 1] , [ 2] in 1976. Cannabispiran and cannabispirone were the names given to the same compound (I)by the Mississippi group [ 1] and by Bercht and others [ 2] . The Mississippi group [ 1] iso1ated cannabispiran from the dried leaves of an Indian variant using a silica gel packed column, while structure elucidation was by x-ray crystallography. Bercht and others [ 2] iso1ated the same compound from the dried leaves of a South African variant grown in France. Structure assignment was based on extensive mass spectral. 1H-NMR and 13C-NMR studies.
Bercht and others [ 2] also isolated cannabispirenone (II)from the South African variant grown in France, while the Mississippi group [ 3] isolated the same compound from the South African variant grown in Mississippi, and called their compound dehydrocannabispiran. An isomeric compound of dehydrocannabispiran with the hydroxy and methoxy groups interchanged (III)was detected by Bosch and Salemink [ 4] in Mexican Cannabisgrown in Mississippi and supplied by the Mississippi group. In l977, the Mississippi group, using the South African variant grown in Mississippi, was able to isolate another related compound, namely, β-cannabispiranol (IV) [ 5] . The β-configuration was assigned, based on the multiplicity of the proton on the carbon carrying the secondary hydroxy group in the 1H-NMR spectrum of the acetate derivative. Comparisons were made with the derivatives obtained by the NaBH 4reduction of cannabispiran. Shoyama and Nishioka [ 6] in 1978 reported the isolation of β-cannabispiranol, which they named cannabispirol; and its monoacetyl derivative (V)together with cannabispiran and dehydrocannabispiran from different strains of Japanese domestic Cannabis,β-cannabispiranol was isolated by the Mississippi group [ 5] one year earlier.
1The term Cannabis in this paper refers to Cannabis sativa L.
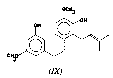
Recently, Crombie and others [ 7] reported the isolation of cannabispiradienone (VI),from Thailand Cannabis.The structure was determined by spectral means and conversion to cannabispiran. The dienone is a possible intermediate in the biosynthesis of spiro compounds and could be formed through phenol oxidative coupling of 3-{2-(4-hydroxyphenyl) ethyl}-5 methoxyphenol. Cannabispiran, β-cannabispiranol and dehydrocannabispiran were also isolated from this variant. Three dihydrostilbenes (VII-VIII)were first reported by Bosch and Salemink [ 4] to be present in the methylene chloride extract of Mexican Cannabissupplied by the Mississippi group. The structure of (VII)was determined by spectral evidence and confirmed by synthesis, while only spectral data were given for (VIII). Compound (VIII) was also isolated from Thailand Cannabis [ 8] along with another dihydrostilbene, called canniprene (IX).The structure was determined by spectral and chemical means and later proved by synthesis [ 9] .
This work describes the isolation and identification of three spiro-compounds (I, II and IV) and two dihydrostilbene derivatives (VIIIand IX) from a Panamanian variant.
Extraction of leaves: 7 kg of the leaves of a Panamanian variant of Cannabis sativa L. grown in Mississippi were exhaustively extracted with 95% ethanol (weight: 1.8 g). The ethanol extract (800 g) was partitioned between chloroform and water. The chloroform phase (fraction A) was partitioned between hexane and cold aqueous solution of NaOH (3N). The basic aqueous phase was rendered acidic with HCl and extracted with ether (weight: 35.2 g, fraction B). Thin-layer chromatography followed by GLC analysis of fraction B showed the presence of several polar cannabinoids. On precoated silica gel G plates it showed four major spots with R f values of 0.31, 0.23, 0.14 and 0.09 using hexane/ether (1:1) as a solvent system. Several minor constituents were also detected with TLC. On polyamide precoated sheets and ethanol/water (7.5:2.5) as a developing system, three spots could be detected with R f values 0.33, 0.56 and 0.7. Furthermore, GC analysis on 2% OV-17 showed major peaks with a relative retention time of 0.33, 0.36, 0.38 and 0.44 in a ratio of 1:1.8:0.5:2, respectively.
Column chromatography of fraction B: Separation of these constituents for identification and quantification was carried out using polyamide column and ethanol water (6 : 4) as the eluting system. The residue (35.2g) was dissolved in chloroform/methanol (8 : 2) and absorbed on acid washed celite (35g). One half of the resulting material was absorbed on column A and the second half on column B using the same conditions as described above.
Column A: A column packed with polyamide (320 g, 90 cm x 4.0 cm) was used. The material was applied on top of the column and elution carried out with ethanol/water (6 : 4). The amount of ethanol was gradually increased. A total of seven fractions were collected. Fractions 1 and 2 (weight 1.4g and 3.48g, respectively) were again rechro-matographed on silica gel G columns.
Column chromatography of fraction 1(1.4 g): GC-MS of this fraction showed three major components having molecular ions at m/e of 248, 244 and 246 with RRT of 0.32, 0.37 and 0.44, respectively, using 2% OV-17 with 4-androstene-3, 17-dione as the internal standard. Therefore, this material was applied on a silica gel G column (160g, 40 cm x 4 cm) packed in hexane/ether (6 : 4). Fractions 23-28 were found to be pure by TLC {R f 0.36 in hexane/ether (1 : 1)} and labelled as compound A. Fractions 51-60 (27mg) were combined and repurified by preparative chromatography using hexane/ether (1 :1) as the developing system. The required zone was located under uv light then scraped and eluted with chloroform to yield a residue (16mg) pure by TLC (R f 0.23) using hexane/ether (1:1) and labelled as compound B. Fractions 200-230 (62 mg) eluted with hexane/ether (1:1) and showing one spot on TLC {R f 0.15 hexane/ether(1:1)} were combined. Crystallization from acetone/hexane yielded sandy crystals (56mg) which were labelled as compound C. Additional amounts of compound A to C were obtained through repeated chromatography of fraction 2 (3.48g) and from another polyamide column (column B) following the same procedure as described under column A.
2Melting points were determined on Koffler's hot-stage apparatus and are uncorrected. The uv spectra were obtained on a Beckman Acta-III spectrophotometer and the ir spectra were determined on a Beckman IR-33 recording spectrophotometer in KBr pellets. 1H1H-NMR spectra were recorded in CDCI 3 on a Jeol-C60-HL instrument with tetramethylsilane as internal standard mass spectra were taken with a Finnigan 3200, MS/DS system. Optical rotations were measured on a Perkin-Elmer 141 polarimeter. Gas chromatographic analyses were carried out using a Beckman GC-65 and 2% OV-17 column following published procedure [ 10] .
Column chromatography of fraction 2 (3.48g):This material was rechromatographed on a silica gel G column (300g, 53.5 cm x 3.3 cm) packed in hexane/ether (6 :4). A total of 10 fractions were collected. Fractions 4 and 7 (1.224 g and 0.278 g) were rechromatographed on silica gel G columns packed in 15% ethyl acetate/cyclohexane. Fraction 4 yielded 172mg identical to compound A through co-chromatography. However, fraction 6 (30.9 mg) showed a major spot of R f 0.42 in 50% ethyl acetate in methylene chloride. This fraction was purified by preparative TLC to give 13mg of needle crystals, designated as compound D.
Column B: The remainder of fraction B was applied on another polyamide column following the same procedure outlined under column A. A total of 9 fractions were collected.
Column chromatography of fraction 1 of column B: Fraction 1 (6.4g) was chromatographed on a silica gel G column (600g, 34 cm x 4 cm) packed in hexane/ether (6:4). Elution was carried out with the same solvent and 22 fractions were collected. Fraction number 10 (473 mg) was then rechromatographed on silica gel G column using 15% ethyl acetate in cyclohexane to give 188 mg of pure material (compound E).
Characterization of Compounds A-E:
Compound A (cannabispiran)(I): Compound A was crystallized from ether/hexane to yield colourless prisms m.p.175°C; IR √KBr/MAX 3370, 3300, 2960, 1690, 1618, 1595, 1505, 1450, 1330, 1295, 1240, 1215, 1195, 1150, 1055, 1035, 940, 835 and 740 cm -1; MS:M+ 246 (15%), 190 (15.3%), 189 (58%), 187 (3.7%), 176 (100%), 174 (32%), 163 (17.7%), 161. (21%), 131 (12.6%), 128 (11.2%), 115 (17.8%), 91 (14.77%), 77 (11.9%); 1H-NMR (CDCl 3) δ 6.38 (d, 1H), δ 6.18 (d, 1H), δ 3.74 (s, 3H), δ 2.92 (t, 2H), δ 2.68 (sextet, 2H), δ 2.53 (sextet, 2H), δ 2.44 (m, 2H), δ 2.22 (t, 2H) and δ 1.83 (m, 2H). The compound was identified as cannabispiran (I)by comparison with GC and other data previously obtained in our laboratory.
Compound B (dehydrocannabipiran) (II) : Compound B was crystallized from ether/hexane as fine needles, m.p. 160°C; IR √KBr/MAX 3200, 3000, 2940, 1645, 1615, 1595, 1505, 1465, 1440, 1330, 1300, 1195, 1140, 1070, 1050, 1030, 925, 850, 830 and 780 cm-1; MS:M+ 244 (30%), 216 (19%), 202 (4%), 201 (12%), 189 (100%), 187 (90%), 176 (2%), 174 (14%), 173 (6%), 163 (5%), 161 (18%), 145 (8%), 144 (12%), 131 (8%), 128 (15%), 115 (30%). 1H-NMR (CDCl 3) δ 6.85 (doublet of doublet 1H, J= 10Hz, J= 1Hz), δ 6.16 (s, 2H), δ 5.80 (d, 1H; J= 10Hz), δ 3.70 (3, 3H), δ 2.90 (m, 2H), δ 2.40 (m, 4H) and δ 2.00 (m, 2H). This compound was identified as dehydrocannabispiran (II)by comparison with GC and other data previously obtained in our laboratory.
CompoundC (β-cannabipiranol) (IV): Crystallization of compound C from ether/hexane afforded sandy crystals, m.p. 179-180°C; IR √KBr/MAX 3430 (sharp), 3250, 2960, 2885, 1615, 1591, 1499, 1365, 1322, 1260, 1200, 1139, 1122, 1095, 1045, 1030, 1015, 960, 890, 830, 800, 740 cm-1: MS:M+ 248 (18%), 230 (10%), 215 (12%), 202 (8%), 201 (6%), 194 (4%), 189 (65%), 187 (20%), 176 (100%), 174 (14%), 161 (18%), 145 (6%), 144 (5%), 131 (10%), 115 (14%). 1H-NMR (CDCl 3) δ 7.95 (s, 1H, exchanges with D 2O), δ 6.10 (s, 2H), δ 3.98 (pentet, 1H), δ 3.60 (s, 3H), δ 3.05 (s, 1H, exchanges with D 2O); the compound was identified as β-cannabispiranol (IV)by comparison with GC and other data previously obtained in our laboratory.
Compound D(3-{2-(3-hydroxy-4-methoxyphenyl) ethyl}-5-methoxyphenol)(VIII): Compound D was crystallized from ether/hexane to yield needles of m.p. 126-127°C, the mass spectrum showed a molecular ion at m/e 274 (32%), a base peak at m/e 138 (100%), and other significant ions at m/e 167 (17%), 122 (74%), 107 (46%) and 94 (72%). The 1H-NMR spectrum (CDCl 3) showed peaks at δ 2.78 (4H, s), δ 3.73 (3H, s), δ 6.31 (3H, m) and 6.76 (3H, m). These data were consistent with those previously reported for 3-{2-(3-hydroxy-4-methoxyphenyl) ethyl}-5-methoxyphenol (VIII)[4, 8].
Compound E (canniprene) (IX): Crystallization of compound E from ether/hexane gave fine needles, m.p. 111°C; IR √KBr/MAX 3360, 3010, 2995, 2960, 2920, 2838, 2820, 1620, 1590, 1580, 1480, 1455, 1440, 1100, 1080, 1060, 1035, 1010, 990, 930, 870, 850, 835, 810, 790, 700, 685, 650 cm-1; MS:M+ 342 (10%), 205 (100%), 191 (15%), 173 (27%), 163 (50%), 161 (17%), 157 (12%), 148 (43%), 145 (30%), 137 (27%), 131 (51%), 114 (16%), 103 (42%), 91 (20%), 77 (23%), 69 (16%); 1H-NMR (CDCl 3); Δ 6.66 (s, 2H, aromatic), Δ 6.26 (m, 3H, aromatic), Δ 5.73 (s, br, exchanges with D 2O), Δ 5.13 (br, t, 1H), Δ 4.93 (s, br, exchanges with D 2O), Δ 3.86 (s, 3H, OCH 3), Δ 3.76 (s, 3H, OCH 3), Δ 3.43 (d, 2H), Δ 2.8 (s, 4H), Δ 1.78 (s, 3H) and Δ 1.70 (s, 3H). This material was identified as canniprene (IX).Canniprene was previously isolated by Crombie and others [ 7] from Thailand Cannabis variant, 16 weeks after germination [ 8] .
The spiro-indan compounds represent a small but important group of natural constituents of Cannabis.These compounds and their related dihydrostilbenes were isolated from different variants, such as Indian, Mexican, South African and Thailand. In this paper, we report on the isolation of these related compounds from a Panamanian variant of Cannabis.Because these compounds exist in small concentrations in the plant material, repeated chromatography was necessary. The plant material was extracted with 95% ethanol and the solvent evaporated. The concentrated ethanol was then partitioned between chloroform and water. The chloroform fraction was then partitioned between hexane and 3N sodium hydroxide solution. The basic extract was then acidified with HCl and extracted with ether. All compounds isolated in this investigation resulted from this acidic fraction. The initial chromatography was carried out using polyamide column and ethanol/water (6:4) as the solvent system and the ethanol proportion was gradually increased. Repeated chromatography of the fractions obtained from the polyamide column resulted in the isolation of three spiro compounds (I, II and IV)and two dihydrostilbenes (VIIIand IX).These compounds were characterized by spectral data (IR, UV, 1H-NMR and MS) as well as by comparison with reference samples. In addition, several minor constituents were isolated and work is in progress for their structure determination. The isolation of two dihydrostilbenes and three spiro-indan compounds from a single variant provides good evidence that the dihydrostilbenes are the natural precursors to the spiro-indan compounds.
A complete evaluation the biological activity of the spiro-indan compounds is under way. The estrogenic properties are of particular interest [ 11] .
Acknowledgement
The authors wish to thank Ms. Mardi Russell and Ms. Cindy Chastain for their technical assistance. Support was given in part by NIDA contract 271-78-3527 and by the Research Institute of Pharmaceutical Sciences.
T. Ottersen and others, "X-ray structure of cannabispiran: a novel Cannabisconstituent", Journal of the Chemical Society Chemical Communications, 1976, p. 580.
002C. A. L. Bercht and others, "Cannabispirone and cannabispirenone, two naturally occuring spiro-compounds", Tetrahedron,vol. 32, 1976, pp. 2939-2943.
003F. S. El-Feraly and others, "Crystal and molecular structure of cannabispiran and its correlation to dehydrocannabispiran, two novel Cannabis constituents", Tetrahedron,vol. 33, 1977, p. 2373.
004J. J. Kettenes-van den Bosch and C. A. Salemink, "Cannabis XIX. Oxygenated 1,2-diphenylethanes from marihuana", Journal of the Royal Netherlands Chemical Society, vol. 97, 1978, p. 7.
005E. G. Boeren and others, "β-Cannabispiranol: a new non-cannabinoid phenol from Cannabis sativa L.", Experientia,vol. 33, 1977, p. 848.
006Y. Shoyama and I. Nishioka, "Cannabis XIII. Two new spiro-compounds, cannabispirol and acetyl cannabispirol", Chemical and Pharmaceutical Bulletin, vol. 26, No. 12 (1978), p. 3641.
007L. Crombie, W. M. L. Crombie and S. V. Jamieson, "Isolation of cannabispiradienone and cannabidihydrophenanthrene. Biosynthetic re1ationships between the spirans and dihydrostilbenes of Thailand Cannabis", Tetrahedron Letters, No. 7, 1979, pp. 661-664.
008L. Crombie and W. M. L. Crombie, "Dihydrostilbenes of Thailand Cannabis", Tetrahedron Letters, No. 47, 1978, pp. 4711-4714.
009L. Crombie, W. M. L. Crombie and S. V. Jamieson, "Extractives of Thailand Cannabis:synthesis of canniprene and isolation of new geranylated and prenylated chrysoeriols", Tetrahedron Letters, No. 21, 1980, pp. 3607-3610.
010C. E. Turner and others, "Constituents of Cannabis sativa L. VIII: Use of silyl derivatives in routine analysis", Journal of Pharmaceutical Sciences, vol. 63, 1974, p. 1872.
011P. W. Wirth and others, "Constituents of Cannabis sativa L. XXI: Estrogenic activity of a non-cannabinoid constituent", Experientia,No. 37, 1981, pp. 1181-1182.


