The Stereochemistry of Morphine
Author: David Ginsburg
Pages: 32 to 46
Creation Date: 1953/01/01
In this review, the fine stereochemistry at the various asymmetric centres in the morphine molecule will be discussed on the basis of degradative evidence. The information gleaned from various synthetic approaches to morphine will also be discussed, in so far as it may elucidate the steric configuration of the alkaloid or of certain of its degradation products. Since it is clearly established that morphine and its methyl ether, codeine, are members of the same stereochemical series, much of the evidence obtained for the configurational relationships in codeine or its degradation products (used primarily because of their greater stability as compared to the corresponding unmethylated compounds) will apply equally to morphine.
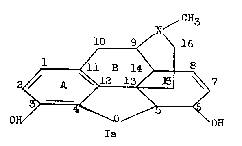
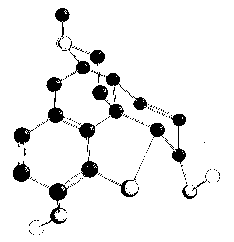
Morphine has five asymmetric carbon atoms: C 5, C 6, C 9, C 13, C 14 (for the numbering system, see structure Ia). The appearance of the molecule in space may be visualized more readily from the above artist's conception. 1
1. The artist's drawing shows the basic skeletal structure of morphine. Most of the hydrogen atoms are not shown. We acknowledge with thanks the co-operation of Mr. Amnon Rubenstein in preparing this drawing.
Emde [1] 2 arranged the atoms of morphine in the manner indicated by structure Ib, in order to emphasize the similarity of the straight chain made up of the five asymmetric centres, to the cyclic forms of aldoses. This formulation is of interest only in that it points out this relationship. It is not useful as a spatial representation of the molecule.
From a study of the optical rotations of certain of the opium alkaloids and their derivatives and employing the principle that the molecular rotation is an additive function of the rotations at the component asymmetric centres, Emde concluded that three of the five asymmetric carbon atoms (C 5, C 6, C 9) rotate the plane of polarized light to the left (-) and the remaining two (C 13, C 14) do so to the right (+). This work shows that the configuration of morphine is the same as in one of the two cyclic forms of aldoses in which the five asymmetric centres are arranged according to the scheme: (-)C 1; (-)C 2; (+)C 3; (+)C 4; (-)C 5, or according to the inverse scheme: (-)C 1; (+)C 2; (+)C 3; (-)C 4; (-)C 5. Although these rotational contributions (+ or -) have been assigned to the asymmetric centres, it must be pointed out that this assignment provides no information as to the actual configurations. Bick (1 a) has recently published a preliminary note in which it is suggested on the basis of rotation displacements that morphine is related to L (+) tartaric acid in its absolute stereochemical configuration.
2. Arabic figures in parentheses indicate items within the list of references to be found at the end of this article.
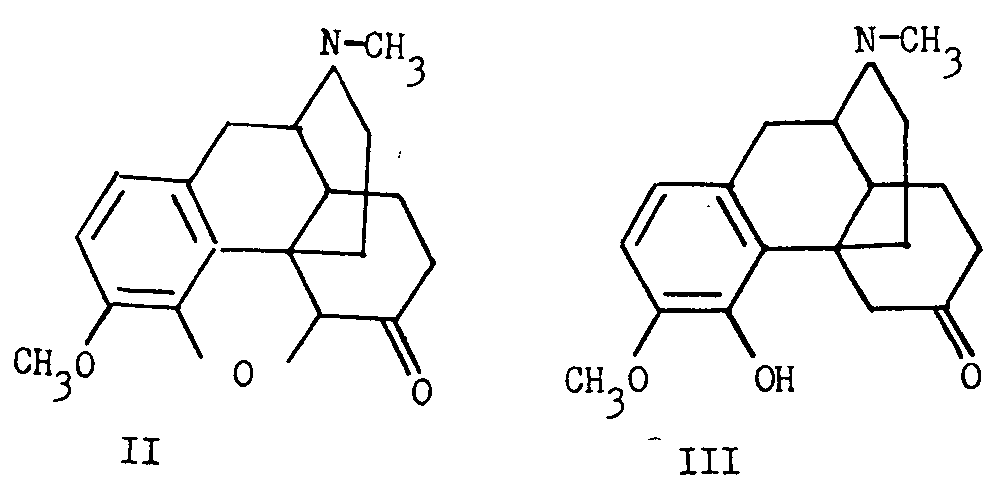
Schopf and Pfeifer [2] , on the basis of a study of models, interpreted the fact that dihydrocodeinone (II)3is formed exclusively from dihydrothebainone (III) by oxide ring closure at C 5, as indicating that the C 5-O bond is transto the ethanamine bond at C 13. The hydrogen at C 14 was inferred to be cis to the ethanamine bond at C 13 by analogy to the situation prevailing in the 14-hydroxy compound. Since degradation of dihydro-14-hydroxycodeinone (VI) to nitrogen-free material resulted in the formation of a cyclic ether (VII) involving the hydroxyl group at C 14, the hydroxyl and the ethanamine bridge must be cisto each other (see chart I).
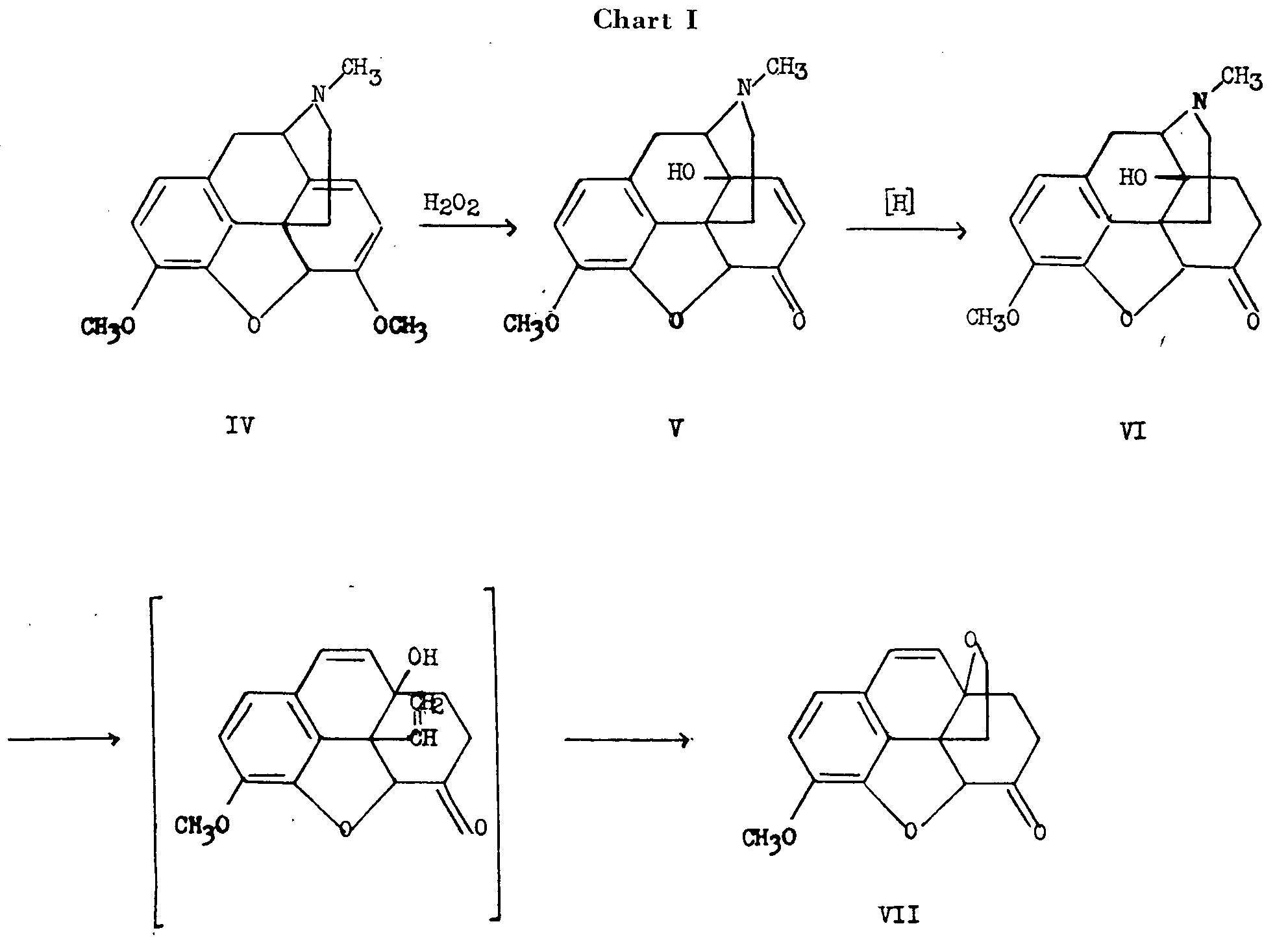
3. Roman numerals in parentheses indicate reference to structures to be found within the charts.
If we assume that the 14-hydroxyl group and the 14-hydrogen in related compounds, possess the same configuration, the hydrogen at C 14 must also be cisto the ethanamine chain.
Gulland and Robinson [3] suggested that hydrogen peroxide causes 1,4-hydroxylation of the diene system of thebaine (IV) with subsequent elimination of methanol to form the C 6 carbonyl group.
There is no ironclad proof showing that the C 14 hydrogen in morphine and codeine has the same configuration' as the C 14 hydroxyl in 14-hydroxycodeinone (V). Schopf assumed that the probability for identical configuration at C 14 in both cases was high because hydrogenation of the 8,14 double bond in various compounds of this series, proceeds in only one steric sense. A study of models indicated that when the hydrogen atom at C 14 is cisto the ethanamine bridge a strain-free system can be obtained. Although compounds having the opposite relationship are known (vide infra), this reasoning, although not affording conclusive proof of the point, was shown to be correct when later work had been carried out by Rapoport and co-workers, who conclusively clarified the configurational relationships of all the asymmetric centres of the morphine molecule.
Rapoport and Payne [4] undertook to determine by absolute methods, the relative configurations at the various asymmetric carbon atoms of morphine.
To determine the relative configuration at C 5 and C 6 the oxide ring was opened at C 4 rather than in the usual manner at C 5 (chart II). A pair of vicinal glycols (at C 5 and C 6) would result from dihydroco-deine (XIa) and dihydroisocodeine (XIb), respec-tively, and their configuration could be determined by usual reagents used for differentiation between cis and trans diols.
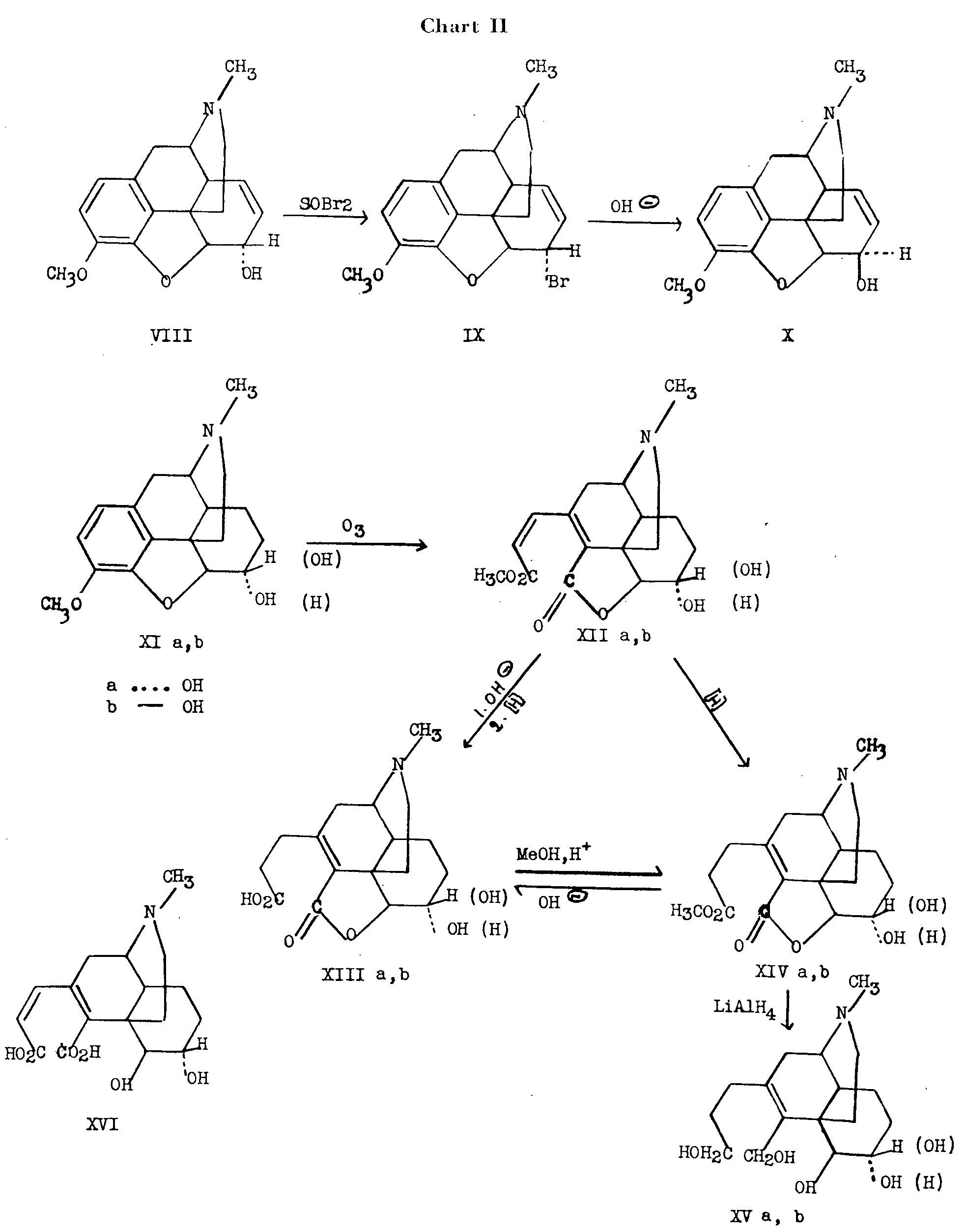
Dihydrocodeine (XIa) was prepared by reduction of codeine (VIII) and dihydroisocodeine (XIb) was prepared in 35 per cent over-all yield by reduction of isocodeine (X), which in turn was obtained from codeine (VIII) by treatment with thionyl bromide to yield bromocodide (IX)4followed by hydrolysis to the alcohol. The transformations carried out with the isomeric pair, dihydrocodeine and dihydroisocodeine are summarized in chart II.
Dihydrocodeine (XIa) and dihydroisocodeine (XIb) differ only in the relative configuration of the hydrogen atom and the hydroxyl group at C 6. In the former, the hydroxyl is represented by the dotted bond (below the plane of the paper) and the hydrogen by the solid bond, while in the latter compound, the reverse relationship holds.
Ozonolysis of dihydrocodeine (XIa) and dihydroisocodeine (XIb), in turn, yielded ozodihydrocodeine (XIIa) and ozodihydroisocodeine (XII) respectively. Hydrolysis of both the methyl ester and lactone groups of ozodihydrocodeine (XIIa) yielded dihydromorphinic acid (XVI), which was too unstable for further work but hydrolysis and reduction of the hydrolysate gave the stable, tetrahydromorphilactonic acid (XIIIa). The latter could be prepared alternatively by first reducing the methyl tetrahydromorphilactonate (XIVa) with subsequent saponification to the acid (XIIIa). The ester could be regained by Fischer esterification.
Tetrahydromorphitetrol (XVa) was obtained by reduction of the morphilactonate (XIVa) with lithium aluminum hydride. Since the C 5-O bond was not broken at any step of these transformations, the configuration at this point remains unaffected and the compound ultimately obtained (XVa) has a pair of vicinal hydroxyl groups at C 5 and C 6 with the original configurations at these carbon atoms retained.
The epimeric tetrol (XVb) was prepared by the same sequence of reactions carried out with dihydroisocodeine (XIb), and the tetrahydro- a-isomorphitetrol (XVb) ultimately obtained, differed from tetrahydromorphitetrol (XVa) merely in configuration at C 6.
It has been shown [5] that cis-1,2-diols are oxidized more rapidly with lead tetraacetate, than the corresponding trans-1,2-diol's.Both tetrols (XVa and XVb) consumed one mole of lead tetraacetate. The α-isomer required more than six hours for consumption of this quantity whereas the other isomer required two hours, i.e., in the dihydrocodeine series the consumption was about thrice as rapid as in the dihydroisocodeine series.
Since an exception to the apparent rule that cisglycols are more rapidly oxidized than their corresponding t ransisomers had been reported [6] , the effect of the glycols on the pH of a boric acid solution was also examined. It has been shown [7] , that the pH of 0.1M boric acid is changed to a greater extent by successive additions of a cis-diolthan by a trans-diol. When this test was applied to the picrate of tetrahydromorphitetrol (XVa) (cisseries) the effect on the pH of 0.1M boric acid was an over-all change in the pH from 5.20 to 4.01 while for the picrate of tetrahydro- α-isomorphitetrol (XVb) transseries), the over-all change was from 5.20 to 4.85.
4. The formulation of IX in chart II is uncertain, as in bromocodide, the bromine atom may be at C 8 and the double bond may be Δ 6 - rather than Δ 7- However, even before unequivocal evidence is furnished regarding this point, the structure of X is certain.
The conclusion was therefore reached by Rapoport and Payne, that in tetrahydromorphitetrol (derived from morphine) the hydroxyl pair at C 5 and C 6 are cisand in the α-isomer (epimeric at C 6) the hydroxyl groups are transto each other. Therefore, in morphine itself, the C-O bonds at C 5 and C 6 are cisand in a-isomorphine they are trans.
Let us now consider the relative configuration at C 6 and C 13. Rapoport [8] has shown that on Hoffmann degradation of dihydrocodeine (XIa), an appreciable amount of methylation of the C 6-hydroxyl group takes place. This observation would indicate the occurrence of intramolecular methylation of the hydroxyl group by the quaternary ammonium ion XVIIIc,d which is an intermediate in the degradation reaction. This led to the idea that if the hydroxyl group at C 6 and the ethanamine chain at C 13 were situated favorably in space, a cyclic ether (XXIII) might be formed as one of the degradation products. Obviously, only the compound in which these groups are cis,would fit the spatial requirements for the formation of the cyclic ether anchored at C 6 and C 13.
Accordingly, dihydrocodeine (XIa) and dihydroisocodeine (XIb), respectively, were subjected to the conditions of the Hoffmann exhaustive methylation and degradation reaction, and the complex mixture of products resulting in each case was separated into a basic fraction (XXIa, XXIb, XXIIa, XXIIb) and a neutral fraction (XIXa, XIXb, XXa, XXb). As will be seen below, the neutral fraction resulting from dihydroisocodeine contained also a small amount of 6-codiran (XXIII). Further, each of these fractions was separated into an alcoholic and non-alcoholic component through formation of the ?-phenylbenzoyl ester of the alcoholic material, followed by purification of this material by sublimation (see chart III).
In the case of dihydrocodeine (XIa) the basic material consisted of the methine (XXIa) and its 6-methoxy-derivative (XXIIa). The neutral material was made up of 6- a-hydroxy-13-vinyl-octahydro-methylmorphenol (XIXa) and the corresponding 6- a-methoxy compound (XXa).
Any cyclic ether (XXIII) would be expected to be present in the non-alcoholic fraction of the neutral material, accompanying XXa. Its absence in this case was shown by quantitative hydrogenation experiments and methoxyl determinations which showed that there was no non-olefinic or non-methoxylated material present. Moreover, in order to separate any suspected saturated material from olefinic compounds, hydroxylation with osmium tetroxide was carried out. The olefinic fraction formed the osmate of the diol (XXIV) which was sufficiently insoluble to effect separation of the olefinic material. This reaction served to confirm the absence of saturated cyclic ether. This evidence indicated that in dihydrocodeine (and therefore in codeine and in morphine), the hydroxyl group at C 6 is trans to the ethanamine chain at C 13.
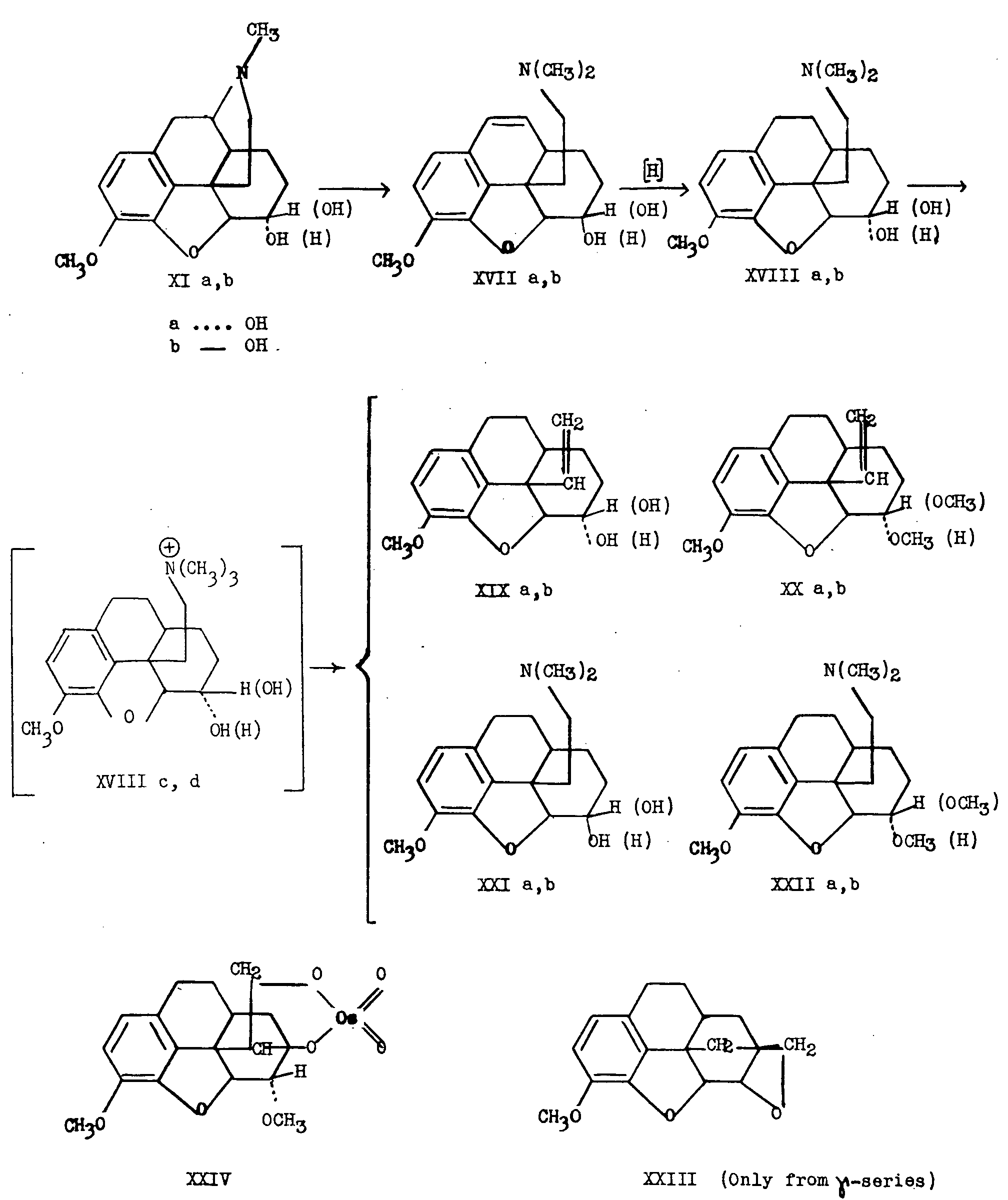
Confirmation was obtained from the results of the reaction sequence carried out with dihydroisocodeine (XIb). A similar mixture of basic and neutral frac- tions was obtained for the 6-γ-isomers. In this case, however, application of the osmium tetroxide hydroxylation procedure separated the vinyl-6-γ-methoxy compound (XXb) from the cyclic 6-codiran (XXIII). It was concluded, therefore, that in the isocodeine series (6-γ-series) the hydroxyl group at C 6 and the ethanamine chain at C 13 are cis with respect to one another.
It is unfortunate that no more than a 2 per cent yield of 6-codiran was isolated from this series of reactions and that the relative proportions of isomeric degradation products were not dramatically different. Thus, it might be expected by the same steric argument used above, that the proportion of methyl ether be greater in the case of the γ-isomer than in that of the α-isomer. The total yield of material unmethylated at C 6 was 64 per cent in the α-series and 34 per cent in the γ-series. Similarly, the total yield of material methylated at C 6 was 34 per cent in the α-series and 61 per cent for the γ-isomers in addition to a 2 per cent yield of 6-codiran in the latter case. The trend is therefore in the expected direction and the assignment in morphine of a C 6-C 13 trans-relationship seems well established.
Returning now to the cis relationship earlier established for the hydroxyl group at C 6 and the C 5-O bond, in morphine, and adding the fact that the C 6-hydroxyl in morphine is trans to the ethanamine chain at C 13, it follows that the hydrogen atom at C 5, the hydrogen atom at C 6 and the ethanamine chain at C 13 must all be cis to each other. A further corollary to the situation prevailing at C 5 and C 13 is the conclusion that the five-membered oxide ring must be fused in the cis manner. The ethanamine bridge joining C 9 and C 13 must be cis since it is impossible in the morphine system to form a six-membered ring across these positions in the trans configuration. Protiva and S?rm [9] have shown that it was impossible to form a lactam from trans-3-amino-cyclohexaneacetic acid but Cronyn [10] and Ginsburg [11] have shown that the cis lactam is readily formed from the isomeric cis-3-amino-cyclohexaneacetic acid or from the ethyl ester of the cis acid.
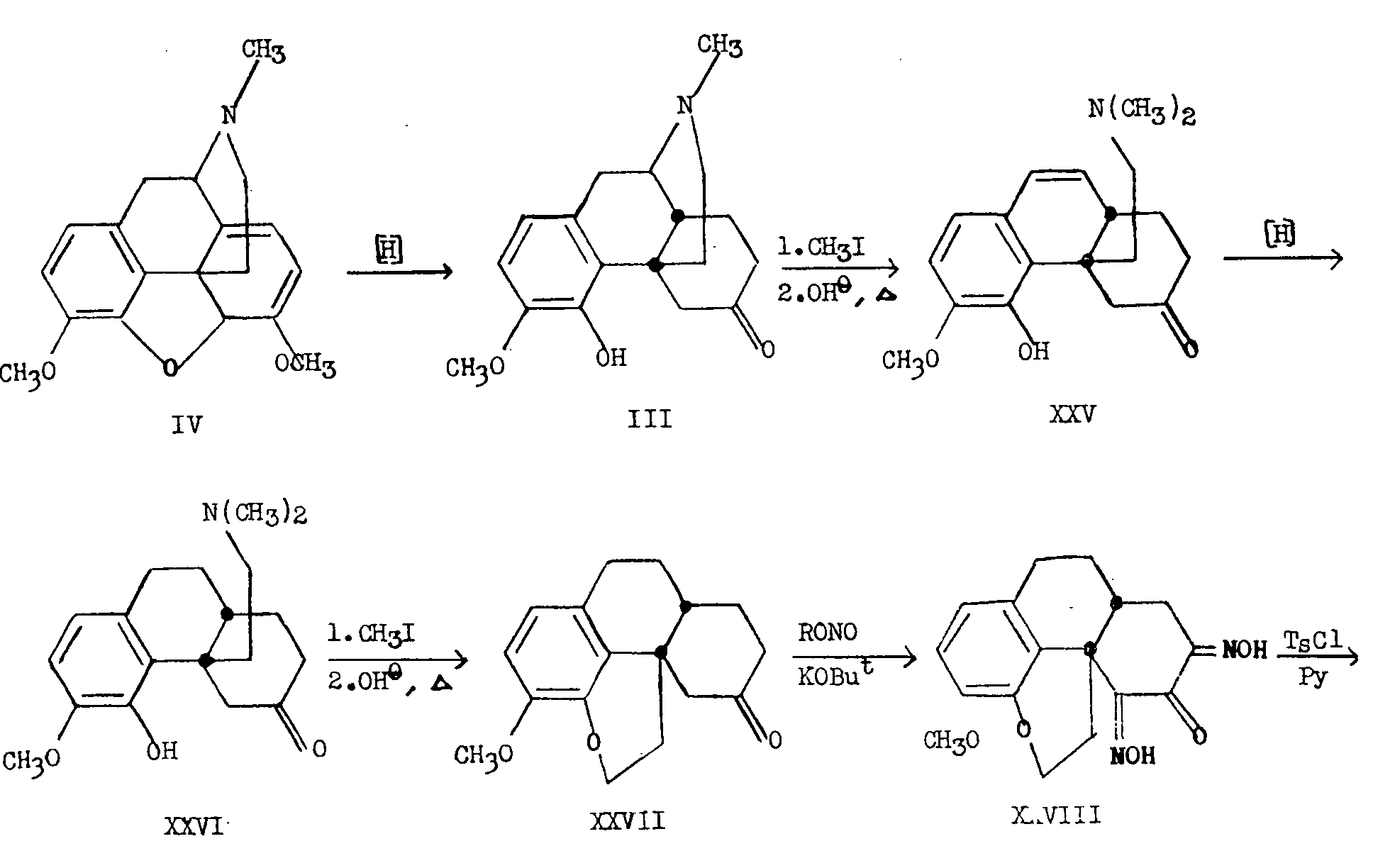
It remained, therefore, to relate the configurations at C 13 and C 14 in order to complete our knowledge about the fine stereochemistry of the morphine molecule. Rapoport [12] has proven the configurational relationship at these centres in the manner summarized in chart IV.
Thebaine (IV) was reduced to dihydrothebainone (III) and after two Hoffmann degradations through the intermediates XXV and XXVI, the 13-vinyl compound presumably formed, yielded the cyclic ether through reaction of the vinyl group with the free hydroxyl group at C 4, so that the product isolated was thebenone (XXVII). Treatment with an alkyl nitrite in the presence of potassium t-butoxide gave 5,7-dioximino-thebenone (XXVIII) which on treatment with ?-toluenesulfonyl chloride in pyridine yielded the dinitrile XXIX. Saponification of the less hindered cyanomethyl group to the acetic acid and further saponification of the hindered nitrile group to the amide was accomplished in stepwise fashion and heating of the resulting monoacid-monoamide XXX, yielded the cyclic imide XXXI since in this series the carboxymethyl and amide groups are in the cis relationship, no reactions having been carried out in the series of transformations which might have permitted inversion at the original centres of C 13 and C 14.
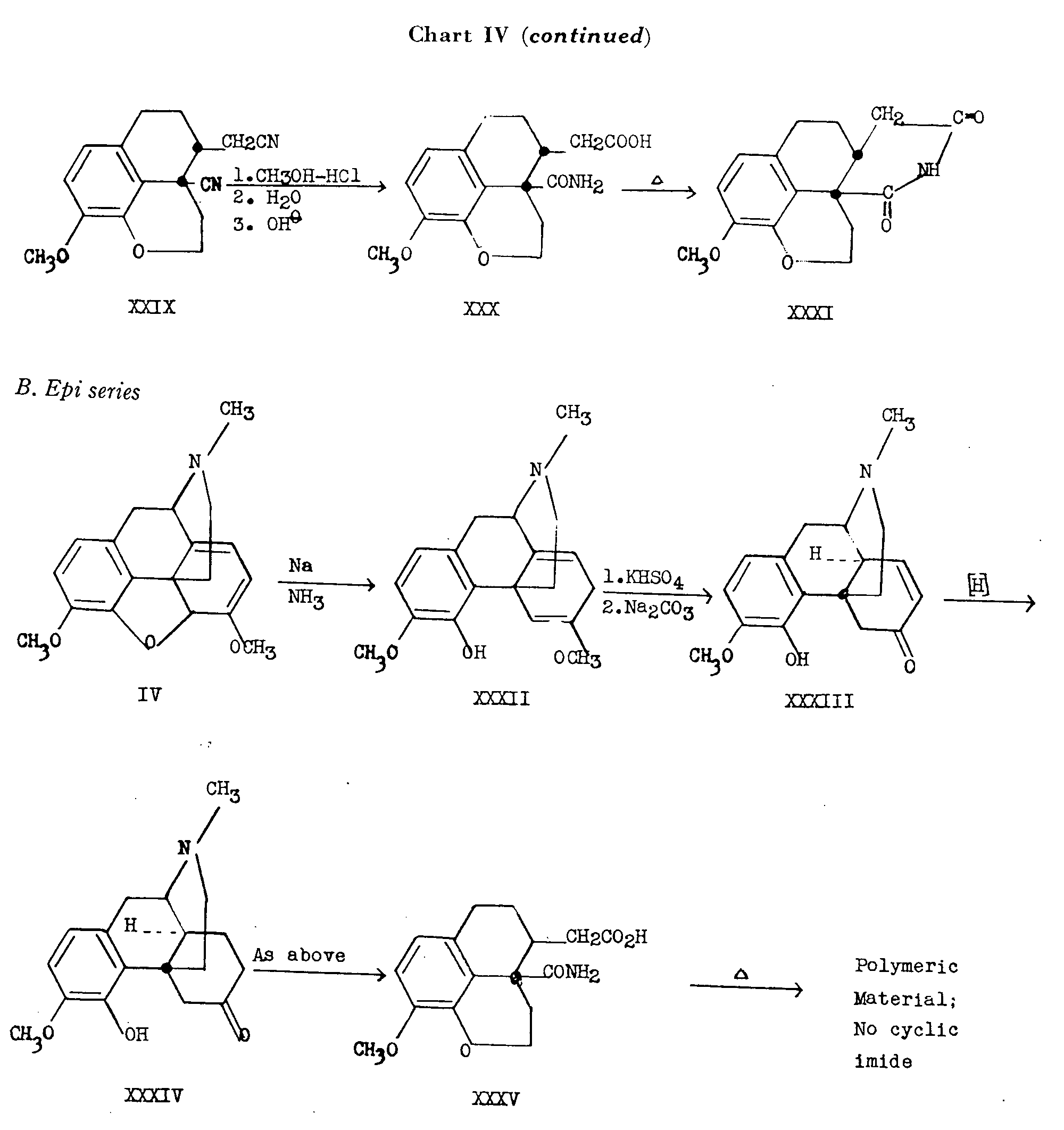
β-Dihydrothebainone (XXXIV), in which rings B and C are locked in the trans-octalin configuration, was prepared by Birch reduction (sodium in liquid ammonia) of thebaine (IV) followed by treatment of the reduction product XXXII with potassium acid sulfate which effects the conversion to β-thebainone (XXXIII). Reduction of β-thebainone yielded β-dihydrothebainone (XXXIV) which was transformed as in the case of the normal series (C 13-C 14 cis), to the corresponding trans-monoacid-monoamide XXXV. When the latter was heated, no cyclic imide was obtained. Only polymeric material was formed.
On the basis of these transformations, it has been shown that the long-presumed cis relationship of the hydrogen atom at C 14 to the ethanamine bridge at C 13, indeed holds true in morphine.
Stork [13] has brilliantly correlated and interpreted a number of other reactions in morphine chemistry in the light of present-day knowledge of the reaction mechanisms involved. These reactions confirm the above steric assignments to the various asymmetric centres of morphine. For further details the reader is referred to Stork's review.
* * *
The synthetic investigations which have been carried out due to the interest in the synthesis of morphine have produced a number of interesting stereochemical points in relation to morphine and certain of its degradation products.
The total synthesis of morphine has been accomlished recently by Gates and co-workers [14] as the culmination of effort expended by many organic chemists for several decades. The synthetic approaches to the morphine molecule have been numerous and have been reviewed by Stern [15] . In this review only a number of stereochemical points omitted in Stern's paper will be discussed.
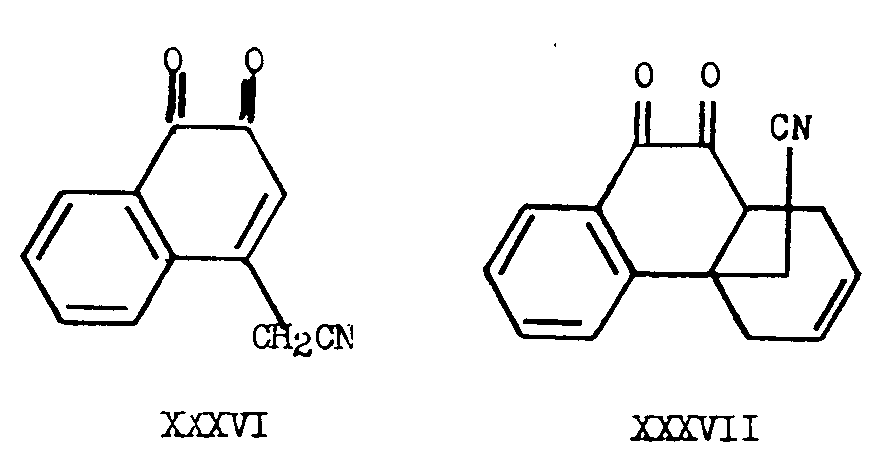
Gates and Newhall [14] prepared trans-4a-cyanomethyl-1, 4, 4a, 9, 10, 10a-hexahydro-9,10-diketophenanthrene (XXXVII) by means of a Diels-Alder reaction of 4-cyanomethyl-l,2-naphthaquinone (XXXVI) with butadiene. Despite the stereospecificity of the Diels-Alder reaction, which would be expected to lead to a cis compound, only the adduct XXXVII in which the hydrogen atom at C 10a and the cyanomethyl group at C 4a were in the trans relationship, was formed, due to the presence of the carbonyl group adjacent to the ring junction, as shown by further synthetic conversion to N-methyl-isomorphinane (XXXVIII) in which rings B and C are trans-locked. In N-methyl-morphinane (XXXIX) prepared by Grewe and co-workers [16] and by Ginsburg and Pappo [17] , rings B and C are cis-locked. These two tetracyclic tertiary amines yield isomeric dihydrodesbases (XLII and XLIII) after Hoffmann degradation and reduction of the resulting desbases (XL and XLI).
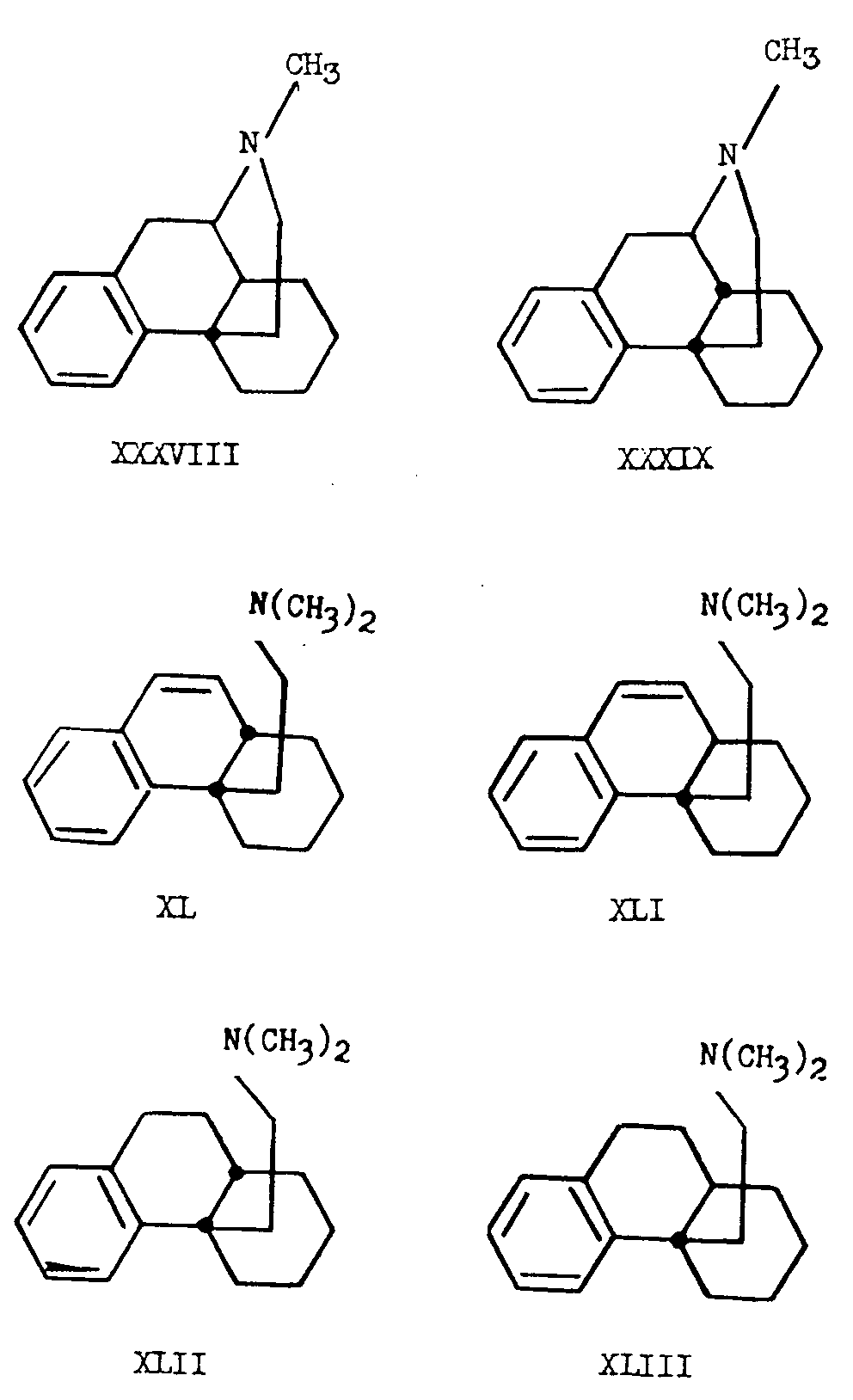
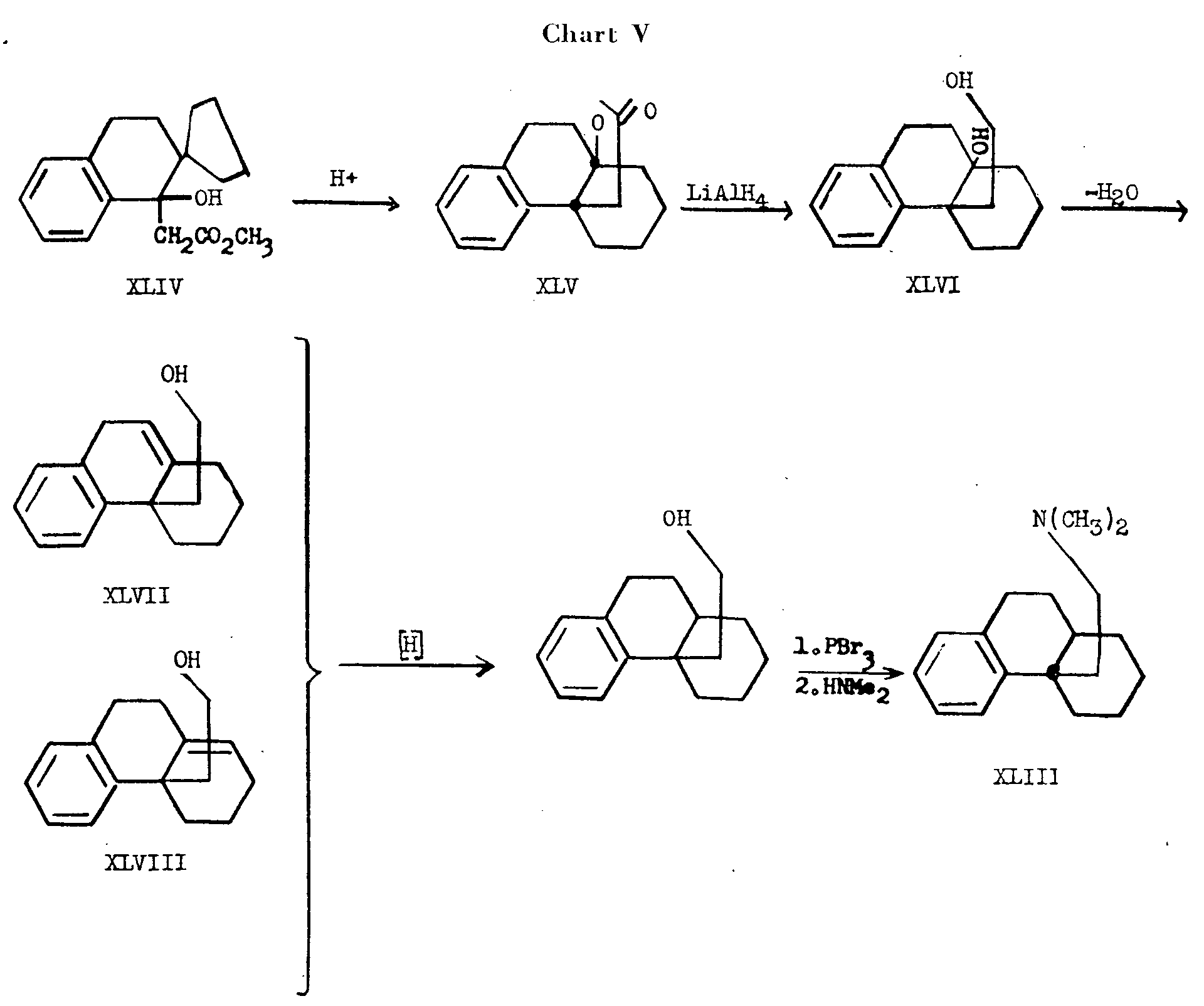
Belleau [18] has synthesized the trans-dihydrodesbase XLIII by Wagner rearrangement of methyl 2-spirocyclopentanotetral-1-ol-1-acetate (XLIV) to the tetracyclic lactone XLV (see chart V). The latter product was reduced with lithium aluminum hydride to give the diol XLVI which could be dehydrated to yield a mixture of unsaturated alcohols (XLVII and XLVIII). Reduction of this mixture followed by conversion to the bromide by means of phosphorous tribromide and reaction with dimethylamine gave a product from which only the trans-dihydrodesbase, trans-4a-β-dimethylaminoethyl-1,2,3,4,4a,9,10,10a- octahydrophenanthrene (XLIII), was isolated in pure form. The product was identical with the dihydrodesbase XLIII obtained by Gates (14) from N-methyl-isomorphinane (XXXVIII).
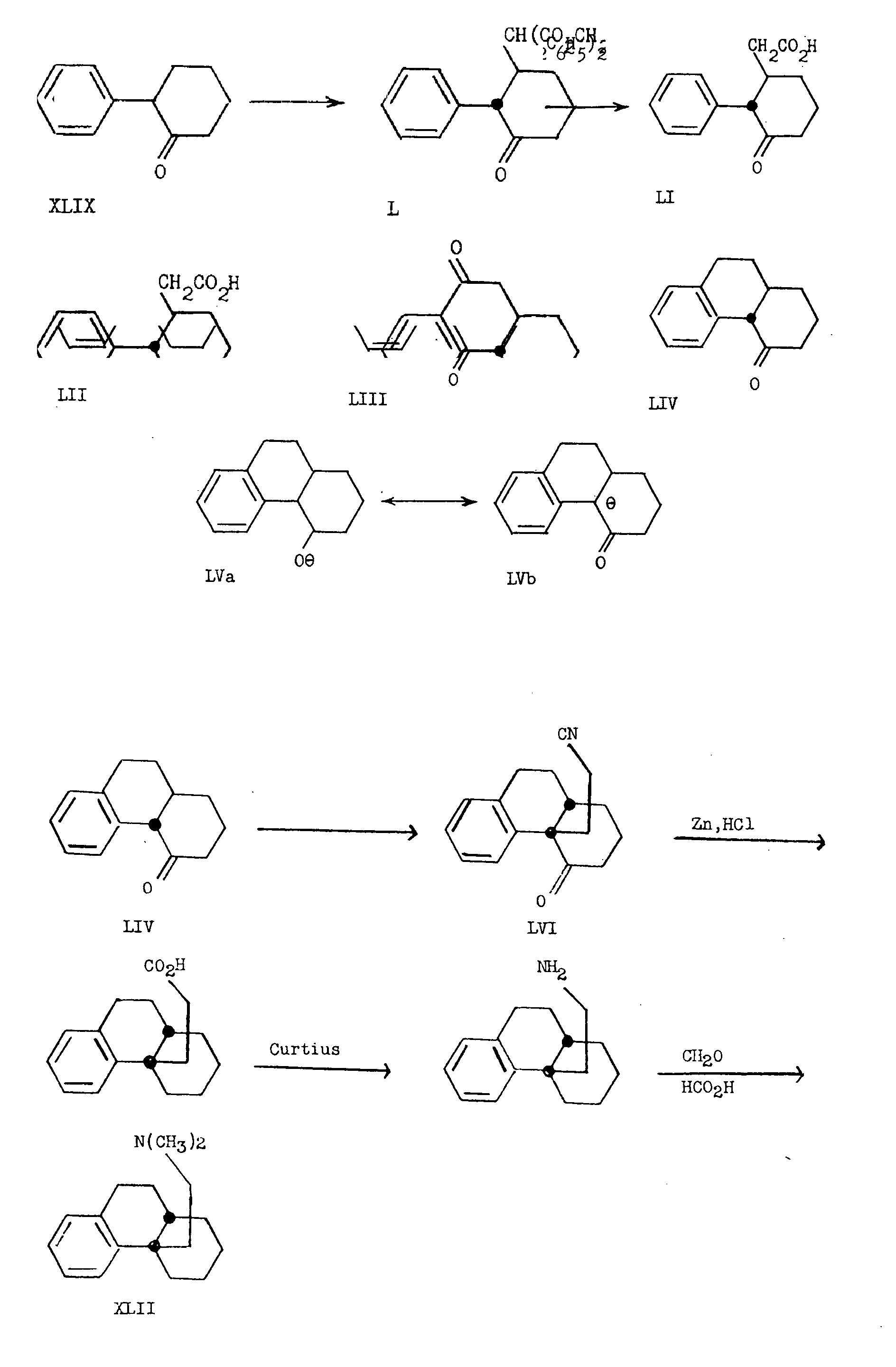
Alternatively, the exclusive synthesis of the cis-dihydrodesbase XLII by Ginsburg and Pappo [17] is of interest, particularly in view of the fact that it was obtained from trans-l,2,3,4,4a,9,10,l0a-octahydro-4-keto-phenanthrene (LIV).
Michael condensation of 2-aryl-cyclohex-2-enones with donors containing reactive methylene groups, proceeds in high yield. For example, 2-phenyl-cyclohex-2-enone (XLIX) with dibenzyl malonate in the presence of potassium t-butoxide affords the adduct L, which upon hydrogenolysis and decarboxylation yields trans-3-keto-2-phenyl-cyclohexane-acetic acid (LI). The configuration of the product was shown by Clemmensen reduction of this keto-acid to the known trans-2-phenyl-cyclohexaneacetic acid (LII). (17, 19). The keto-acid (LI) can be cyclized to yield trans-l,2,3,4,4a,9,10,l0a-octahydro-4,9-diketophenanthrene (LIII) in a reaction which still maintains the existing stereochemistry at C 4aand C 10a. Chart VI summarizes these reactions and the further utilization of LIII in the synthesis of the cis-dihydrodesbase (XLII).
Cyanoethylation of the trans-monoketone LIV, obtained by catalytic reduction of the trans-diketone LIII, yields exclusively the cis-4a-β-cyanoethyl derivative LVI. This product was converted to the cis-dihydrodesbase, cis-4a-β-dimethylaminoethyl-1,2,3,4,4a,9,10,l0a-octahydrophenanthrene by a series of simple transformations shown in chart VI. The cyanoethylation reaction which converts a trans- locked octalin system to one having the cis-juncture can be readily understood from the mechanism of the Michael condensation which is believed to proceed through the enolate ion (LVa) or the carbanion (LVb) in which the trans configuration no longer obtains. It is evident that under the alkaline conditions of the Michael condensation, the cis-alkylation product is more stable than the trans-isomer.
Another stereochemical point of interest in this field concerns the preparation of N-methyl-morphinane (XXXIX) from the intermediate LVII. The reaction sequence involved is summarized in chart VII.
The 4-ethylene glycol ketal of trans-l,2,3,4,4a,9,10,10a-octahydro-4,9-diketophenanthrene (LVII) was treated with n-amyl nitrite in the presence of sodium ethoxide to yield the 10-oximino-derivative LVIII. Catalytic reduction of this compound in the presence of hydrochloric acid afforded two isomeric amine hydrochlorides (LIX) and (LX), presumably differing in configuration at C 10a. The more abundant of these (LX) was treated with the acid chloride of acetylglycollic acid to give the amide LXI, which on attempted ketalization, surprisingly cyclized to form the diketolactam LXII. The latter was converted, as shown in chart VII, to N-methylmorphinane (XXXIX) having the cis-octalin configuration. Here again, we are dealing with a case in which the technique cyclization takes place in only one steric sense. Although the mechanism of the cyclization reaction is not known, the lactam of the morphinane rather than the isomorphinane configuration, is the exclusive product. Since only epimerization of the hydrogen atom at C 4a is possible in LX and a cis-ring junction is obtained in the resulting lactam, it may be deduced that LX has the structure shown, in which the amino group at C 10 is equatorial. This would be expected to be the stable conformation of the amino group in a compound with trans-ring junction.
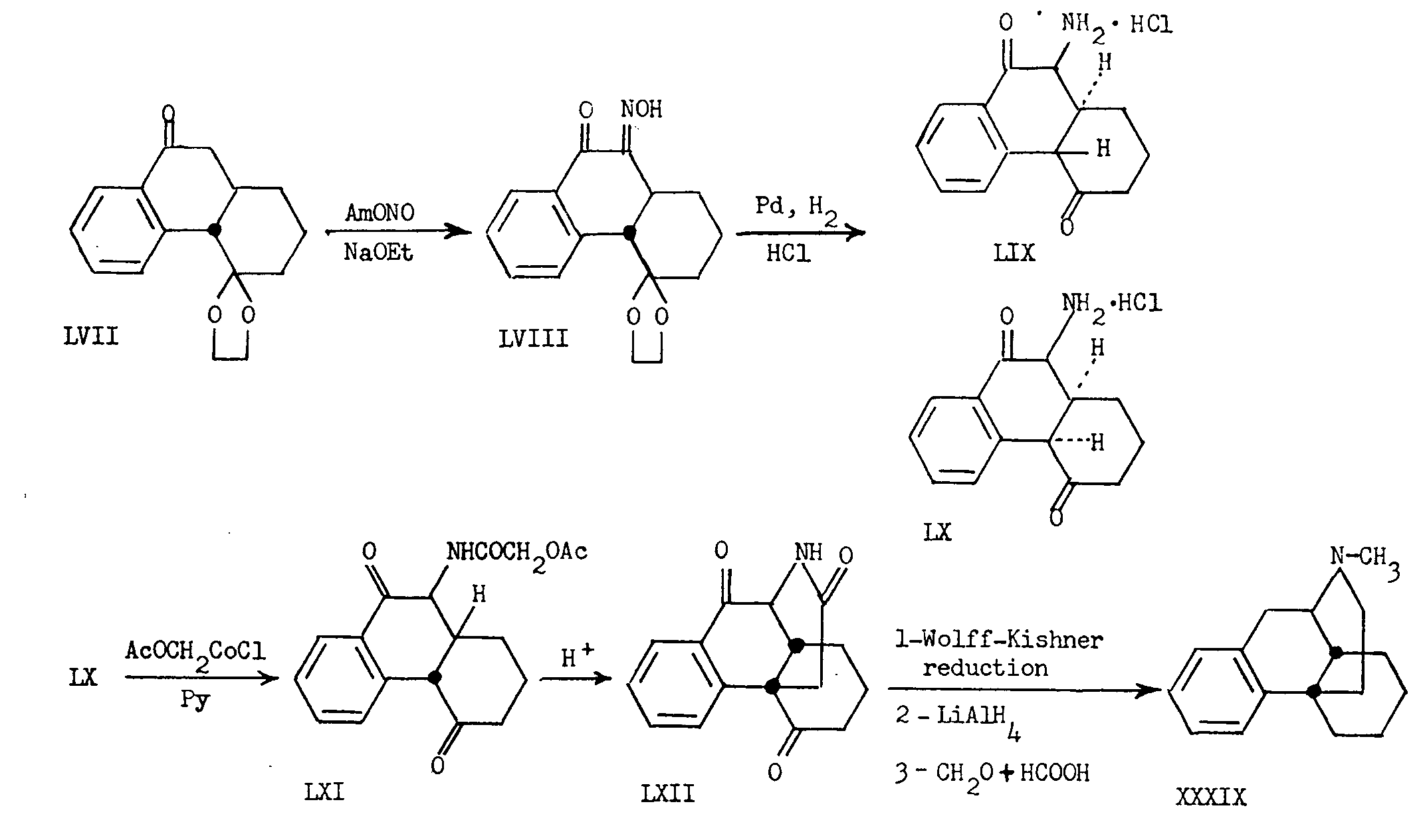
Also worthy of note, stereochemically, is the cyclization reaction discovered by Grewe [16] in which a product of preponderantly cis configuration is formed at C 13-C 14.
In 1928, Robinson [19] proposed a possible biogenetic relationship between the morphine group of alkaloids and those of the 1-benzylisoquinoline group such as laudanosine (LXIII). The biogenetic conversion of a completely demethylated laudanosine precursor (LXIV) to morphine, requires the formal addition of two hydrogen atoms followed by the loss of the elements of one molecule of water. Robinson's hypothesis is extremely attractive since laudanosine (LXIII) can be formed by simple methylation of the precursor LXIV and the conver- sion of the latter to morphine involves reactions, for which most probably, enzyme systems exist in the plant.
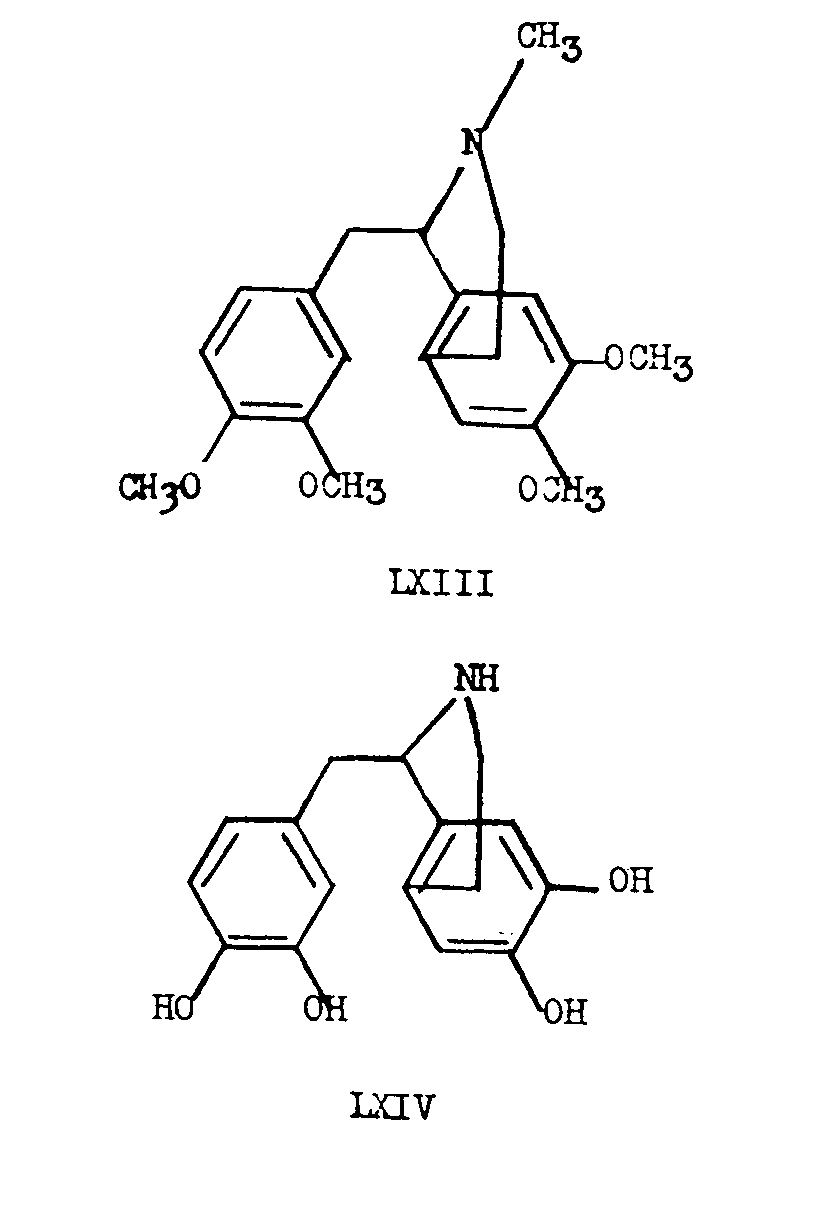
Chart VIII
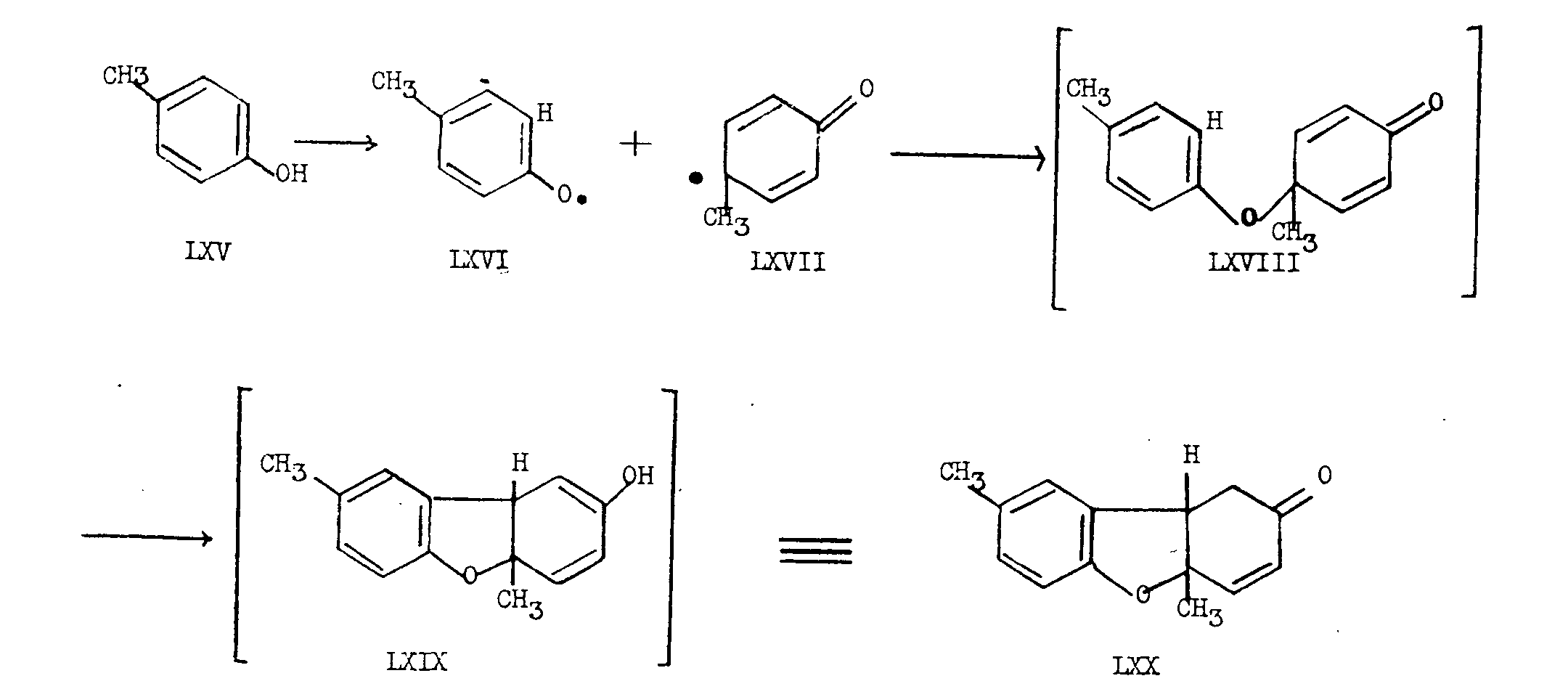
Schopf has recently published a modified biogenetic scheme which relates the morphine alkaloids to the 1-benzyl-isoquinoline group [20] . This scheme is based on the analogy to the dehydrogenation reaction, investigated by Pummerer and co-workers [21] , in which p-cresol (LXV) is converted to the angularly substituted furan derivative LXX, presumably as shown in chart VIII.
In this formulation, it is suggested that p-cresol (LXV) gives a radical which reacts partly in the aryl-oxy form LXVI and partly in the γ-keto-methyl form LXVII, so that when the free valences unite to give LXVIII the hydrogen atom ortho to the oxygen in the aryloxy portion of LXVIII adds intramolecularly 1,4 to the ends of the enone system in the γ-keto-methyl portion to give the intermediate LXIX. LXIX is immediately transformed to the stable LXX by ketonization.
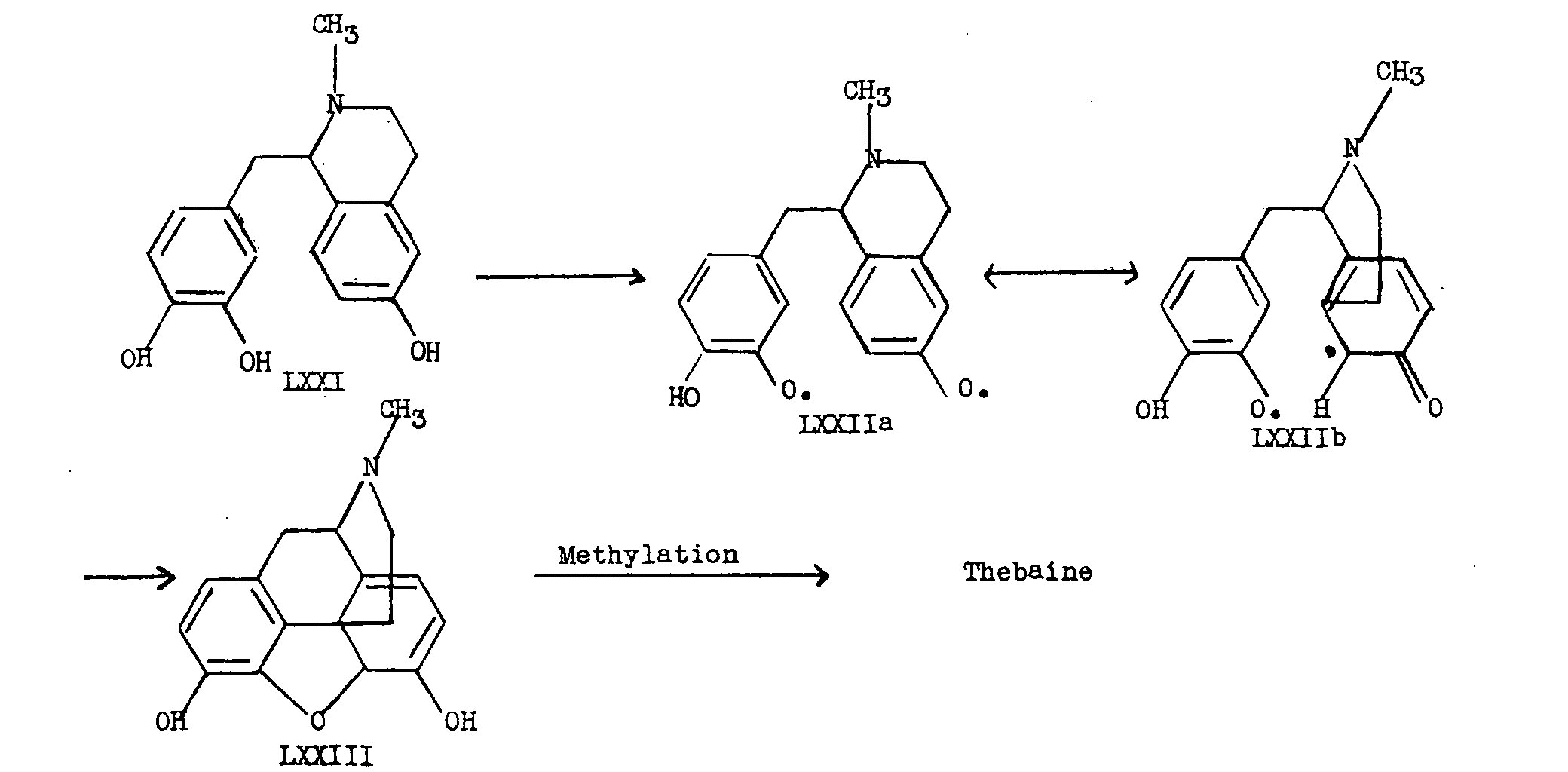
Analogously, Schopf visualizes an intermediate LXXI differing slightly from Robinson's precursor, which must be dehydrogenated to the biradical LXXIIa or LXXIIb. In this case, when the free valences unite intramolecularly, we obtain by 1,6-addition to the ends of a dienone system which differs from the p-cresol case in that it contains an α-keto-methyl system (see LXXIIb), the hypothetical precursor LXXIII. This formulation is summarized in chart IX. It will be seen from chart IX that the precursor LXXIII can very easily afford morphine, codeine, thebaine or neopine (LXXIV).
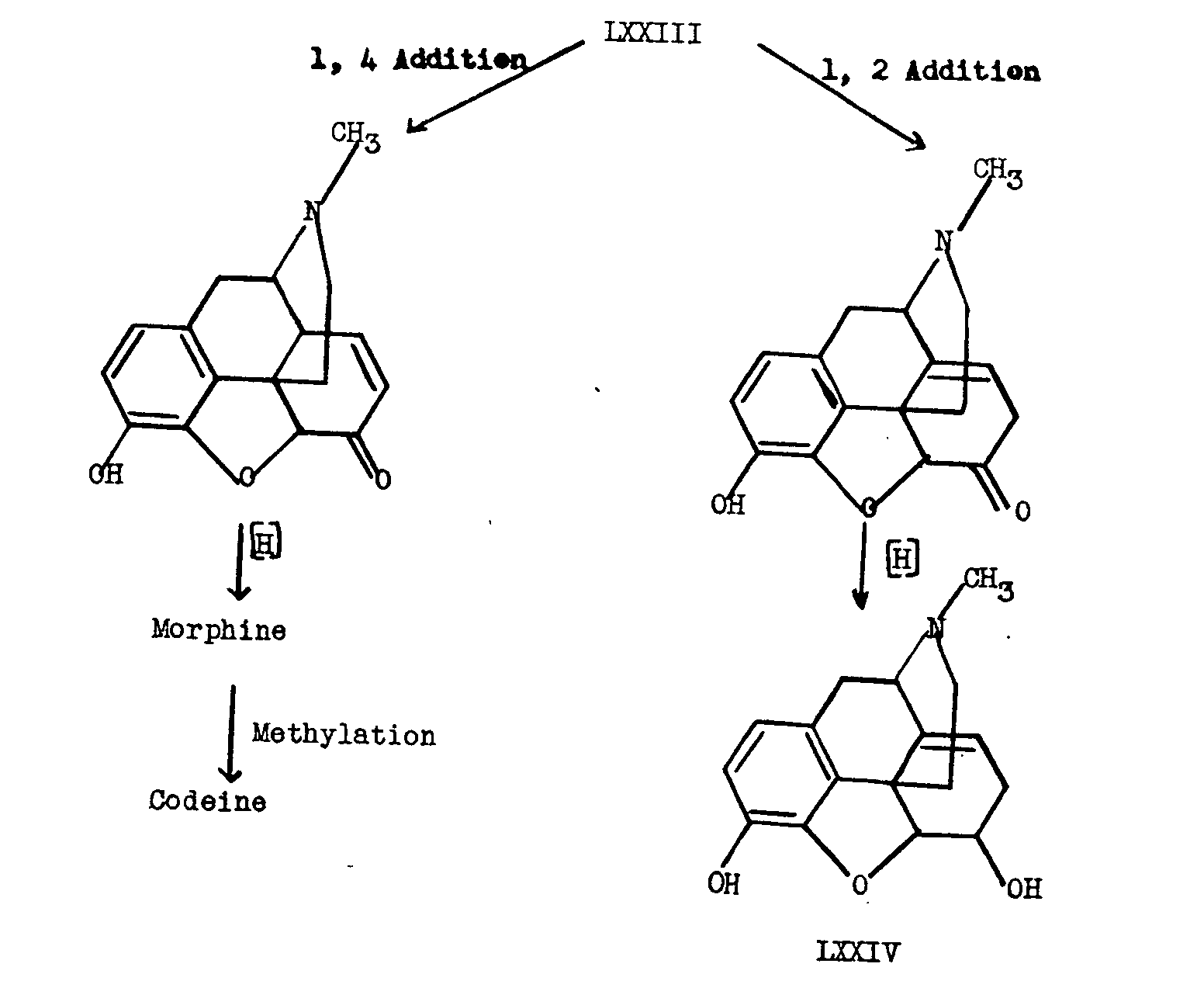
For the difficulties involved in the experimental testing of this biogenetic hypothesis, the reader is referred to Schopf's paper [20] .
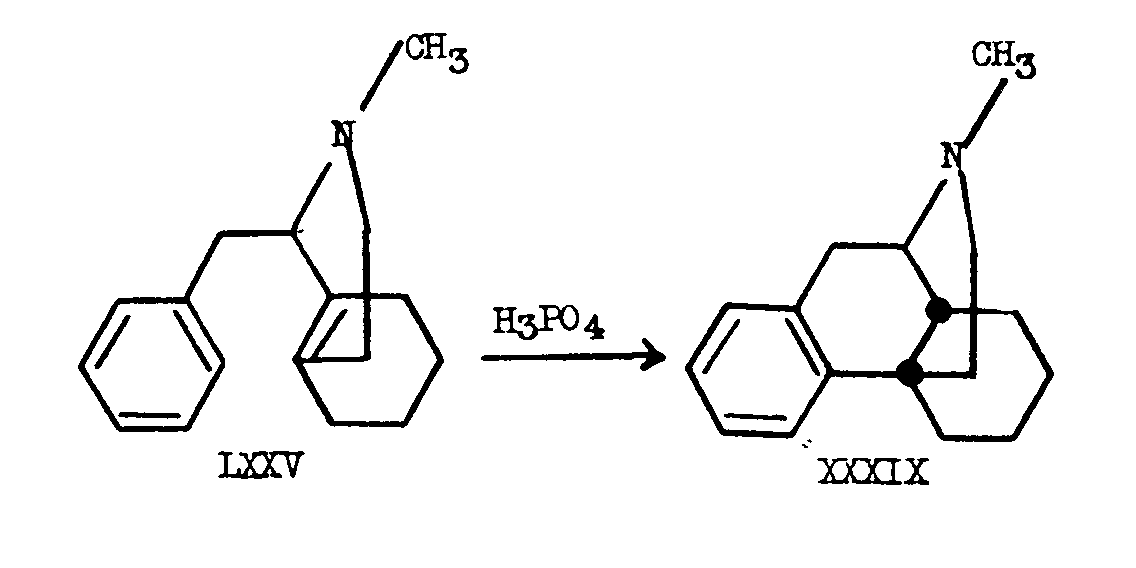
In all the synthetic approaches to morphine, the nearest to the presumed biogenetic route, is that of Grewe [16] . Although this approach culminated in the very elegant and unique cyclization in which the morphine skeleton was formed from a 1-benzyl-octa- hydroisoquinoline, the reactions were not carried out under simulated physiological conditions. The preponderantly stereospecific synthesis of N-methylmorphinane from 1-benzyl- N-methyloctahydroisoquinoline (LXXV) by Grewe produced this compound in 50 per cent yield. A picrate of another isomeric substance was formed only in small amount. It was later shown by Gates [14] that this substance was identical with the picrate of N-methylisomorphinane.
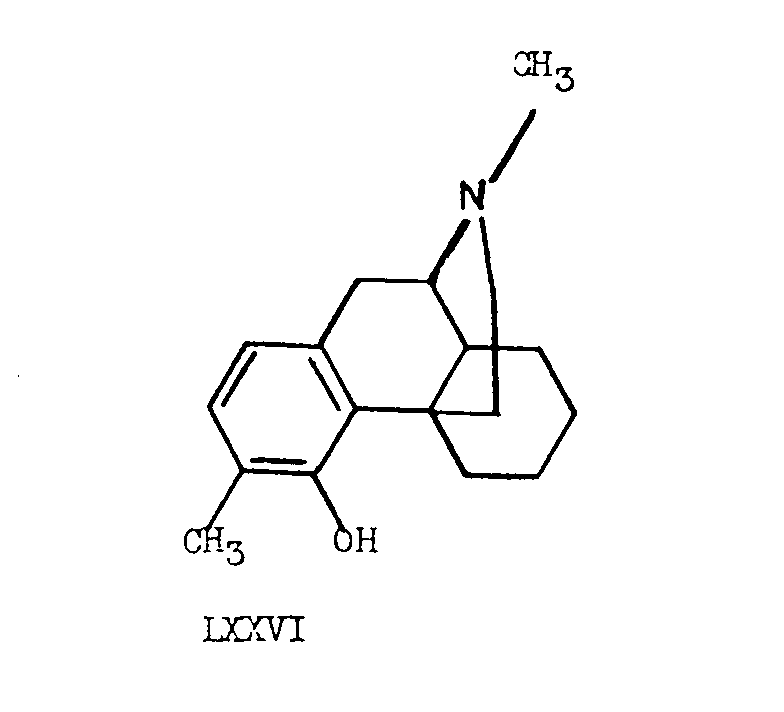
Tetrahydrodesoxycodeine (LXXVI), identical with that obtained from natural sources was also prepared by Grewe and his co-workers [16] , employing the same acid cyclization method in which the addition of a hydrogen from the aromatic ring across the tetra-substituted double bond took place to yield the cis-octalin structure prevailing in the natural product.
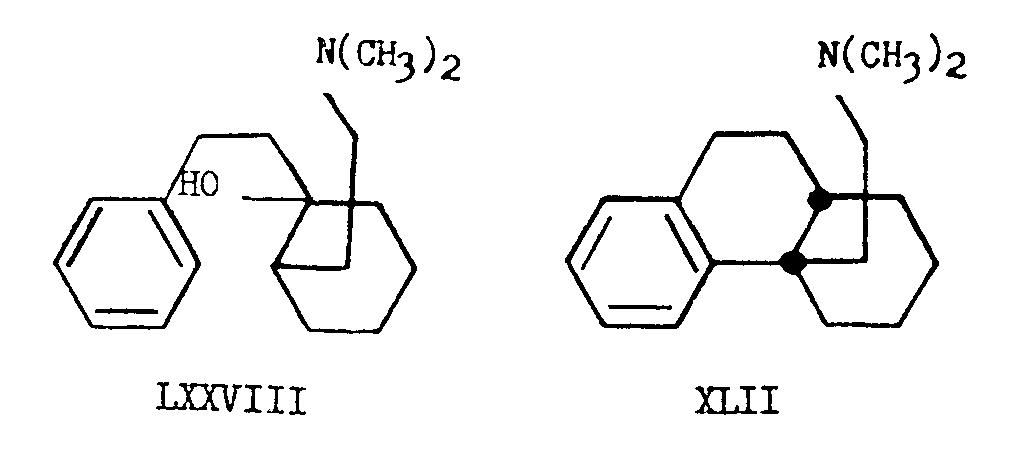
It is clear that in order to obtain the natural, cis-octalin, the cyclization mechanism must involve trans addition of the hydrogen atom to the tetra-substituted double bond. In other known cases, such as the cyclization of β-phenethylcyclohexylene (LXXVII) mixtures of cis and trans products are obtained [22] . This apparently depends upon the stability of the carbonium ion intermediates involved in this type of cyclization and it is noteworthy that despite the fact that there appears to be no great difference in stability when Fisher-Hirshfeld models of N-methylmorphinane and N-methylisomorphinane are studied, the cis isomer is formed in this case preponderantly. Analogously, the cis-dihydrodesbase (XLII) could be obtained by treatment of the amino-alcohol LXXVIII with syrupy phosphoric acid which effects dehydration followed by cyclization.
The final steric point which must be discussed is the remarkable isomerization of the trans-C 13-C 14 system to the isomeric cis system during the course of Gates' work on the total synthesis of morphine. The reactions involved are summarized in chart X.
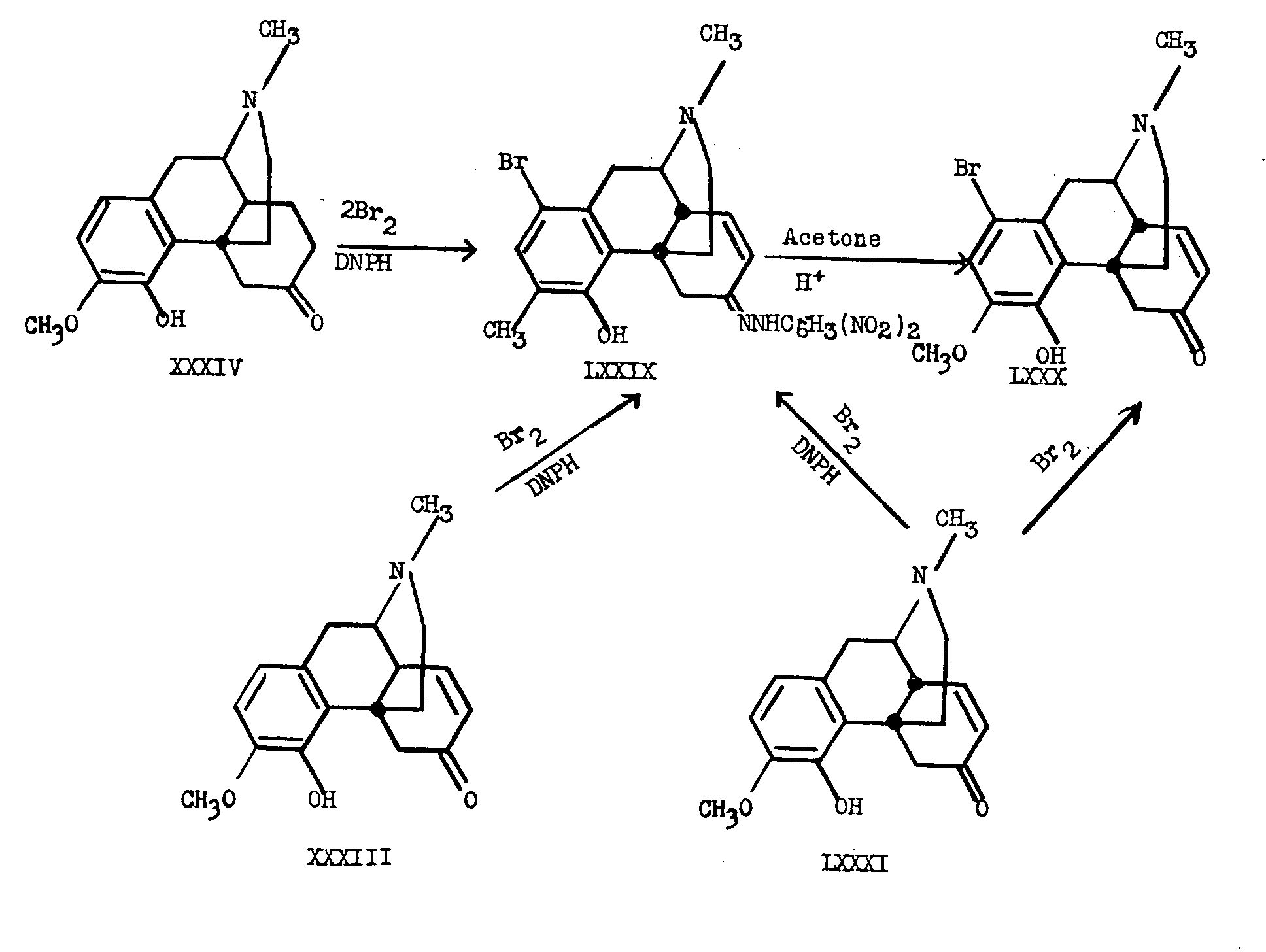
When β-dihydrothebainone (XXXIV) was brominated with two moles of bromine followed by treatment with 2,4-dinitrophenylhydrazine, a dinitrophenylhydrazone LXXIX, was obtained. This derivative was identical with the 2,4 dinitrophenylhydrazone which results from β-thebainone (XXXIII) or from thebainone (LXXXI) by the action of 2,4-dinitrophenylhydrazine in acetic acid with subsequent bromination.
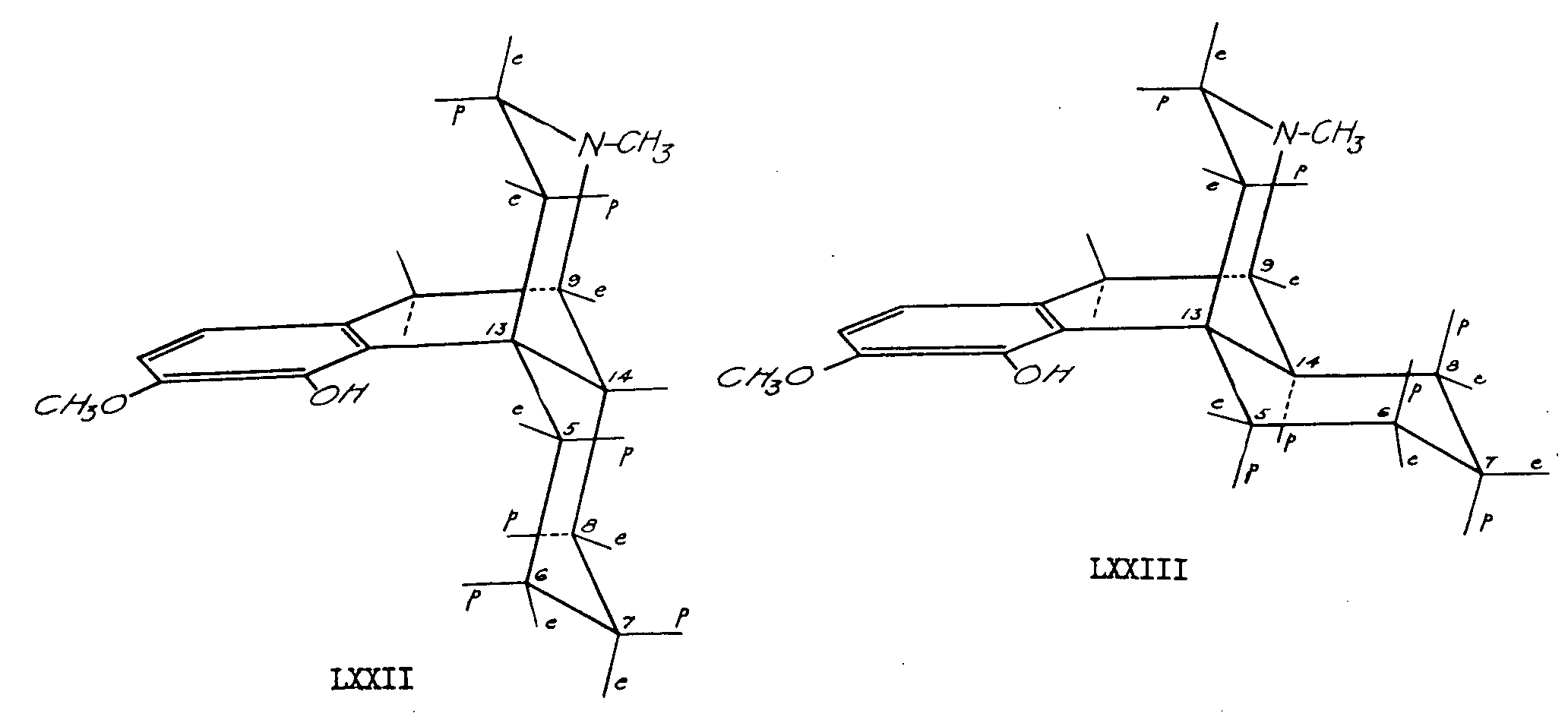
The epimerization at C 14 took place so readily that the 2,4-dinitrophenylhydrazone of β-thebainone could be obtained only by minimizing contact of the compound with acid. The abnormally high rotation of Δ 7-6-ketone-dinitrophenylhydrazones of the cis series as compared to that of derivatives of the trans series could be used to follow the progress of the epimerization. The difference in molecular appearance of cis and trans compounds of this type is illustrated in structures LXXXII and LXXXIII which are reproduced through the courtesy of Dr. Marshall Gates.
It may be seen that each of the approaches to the morphine structure has produced some unique or remarkable reaction. As in the case of many natural products, the interest in morphine has led to the development of new methods and reactions of interest to chemistry as a whole.
The variety of rearrangements which morphine undergoes has earned for it, in the words of Sir Robert Robinson [23] , the reputation of being "a star performer among molecular acrobats".
It is hoped that this article has shown that morphine, possibly to a greater extent than any other single natural product, has created extensive interest among organic chemists. Those chemists who have, in whole or in part, successfully attacked the problem of its synthesis, have been more than once rewarded, perhaps because of the elusiveness of the "star performer" with rich gifts of stereochemical good fortune by a merciful and bounteous Providence.
This article is dedicated to Dr. Lyndon Frederick Small whose work on opium alkaloids contributed in large measure to the author's interest in this field.
Emde, Helv. Chim. Acta, 13, 1035 (1930).
02Bick, Nature, 169, 755 (1952).
03Schopf and Pfeifer, Ann., 483, 157 (1930).
04Gulland and Robinson, J. Chem. Soc., 123, 980 (1923). Cf. Freund and Speyer, J. prakt. Chem., 94, 135 (1916).
05Rapoport and Payne, J. Org. Chem., 15, 1093 (1950).
06Criegee, Ber., 64, 260 (1931); Criegee, Kraft and Rank, Ann., 507, 159 (1933).
07Boyland and Wolf, Biochem. J., 47, 64 (1950).
08Boeseken, Advances in Carbohydrate Chemistry, vol. IV, Academic Press, New York, 1949. p. 189.
09Rapoport, J. Org. Chem., 13, 714 (1948).
10Protiva and S?rm, Coll. Czech. Chem. Comm., 13, 428 (1948); Chem. Abstr., 43, 1730 (1949).
11Cronyn, J. Org. Chem., 14, 1013 (1949).
12Ginsburg, ibid., 15, 1003 (1950).
13Personal communication from Dr. Henry Rapoport.
14Stork, The Alkaloids, vol. II, Academic Press, New York, 1952, pp. 171-189.
15Gates, et al., J.A.C.S., 70, 2261 (1948); ibid., 72, 1141, 4839 (1950); Experientia, 5, 285 (1949); J.A.C.S., 74, 1109 (1952).
16Stern, Quart. Rev., 5, 405 (195l).
17Grewe. et al, Ber., 81, 279 (1948); ibid., 84, 527 (1951); Ann., 564, 161 (1949).
18Bergmann, Pappo and Ginsburg, J. Chem. Soc., 1950, 1369; Ginsburg and Pappo, ibid., 1951, 516, 938.
19Personal communication from Dr. B. Belleau.
20Robinson. Proc. Univ. Durham Phil. Soc., 8, part 1, 14-59 (1927-1928).
21Schopf, Naturwiss., 39, 241 (1952).
22Pummerer et al., Bet., 58, 1808 (1925)
23Cook, Hewett and Robinson, J. Chem. Soc., 1939, 168.
24Robinson, Proc. Roy. Soc., London, B 135, XIV (1947).

