I. ISOLATION AND SEPARATION OF ALKALOIDS
II. DETERMINATION OF INDIVIDUAL ALKALOIDS
III. CALCULATION OF RESULTS
IV. DETERMINATION OF MORPHINE IN ISOLATED POPPY CAPSULES OR PARTS OF SAME
Summary
Author: S. Pfeifer
Pages: 18 to 33
Creation Date: 1958/01/01
Morphine being one of the most important alkaloids used in medicine; it is only natural that methods for the quantitative determination of morphine in opium, in the poppy and in galenical preparations should occupy an important place in the literature on alkaloid analysis. Descriptions of methods for the separation and determination of the by-alkaloids in the poppy and in opium, by contrast, are far less numerous. Moreover, these descriptions concentrate mainly on opium, and could not be applied as they stand, since far smaller amounts of these alkaloids occur in the poppy. Methods for the quantitative determination of the main alkaloids in opium or in total-alkaloid preparations have been described, inter alia, by E. Anneler (1), B. Kljatschkina (2), and D. C. Adamson, F. P. Handisyde & H. W. Hodgson (3). A spectrophotometric method for investigating Pantopon [proprietary name] has also been published by M. S. Dyer & A. J. McBay (4), but this is so far suitable only for pure-alkaloid mixtures. As far as we know, no method of measuring the codeine, thebaine, narceine, narcotine, papaverine, and narcotoline content, as well as the morphine content of the poppy plant, has yet been elaborated. The few authors to have reported at all on the by-alkaloid content have determined only the total by-alkaloid content, or have isolated these alkaloids from large quantities of drugs. Thus, W. Kussner (5), in his interesting experiments, used 10 g of non-phenolic alkaloids in each case and separated them by a series of analyses based on a modified form of Anneler's method. To obtain these 10 g of alkaloids, some 10 kg of drugs is required. A procedure of this kind is quite unsuitable for serial experiments - e.g., for following the alkaloid development in growing plants. There seemed, therefore, to be two good reasons for devising a method for the quantitative determination of the most important alkaloids in the poppy which entailed only slight expenditure of materials and required only a reasonable amount of time - namely, (1) the classification of types of poppy by alakloid groups, for cultivation purposes;[1] (2) .determining the alkaloids during growth, in order to ascertain the biogenetic connexions of the poppy alkaloids.
In this connexion, it should be noted that, despite a number of works dealing with the qualitative or quantitative development of the alkaloids in the poppy plant, research into the biosynthesis of these alkaloids is still in its infancy. Moreover, research to date, which has dealt mainly with morphine and has often been determined by considerations of plant cultivation, has produced very conflicting results. Thus, according to A. Malin (8), the morphine content decreases as the poppy capsules ripen, whereas the codeine and narcotine contents allegedly increase. L. Fuchs (9) and, more recently, A. Guillaume & J. Faure (10) have made similar observations, the two last-named authors postulating that the oil and the morphine occur in inverse proportion to each other. A. Müller (11), reached the same conclusion with regard to the total alkaloid content. H. Baggesgaard-Rasmussen & O. Lanng (12) found that the morphine content varied with the climate and the time of the harvest. W. Kussner (loc. cit.), E. Wegner (13), and J. Tomko (14), however, found that the morphine content rose continuously until the capsules ripened. These studies are, however, again contradicted by a recent publication by M. Kucera (15), who found that the morphine content decreased as the plant ripened. W. Poethke & E. Arnold (16) who, in a model series of observations, followed the morphine content in the individual parts of the plant during growth, explained these differences by outside influences, particularly that of the weather; one year they found the highest morphine content in fully ripened capsules, the following year in the unripe capsules. Generally speaking, this is confirmed by our own research covering several years. J. C. Jespersen (17), I.v. Kabay (18), and H. M. Wüst & A. J. Frey (19) have also given data on the morphine content of leaves, head, and stems. According to E. Wegner (20), the morphine originates in the root. Opinions are also very much divided as regards the increase or decrease of alkaloids during storage. Our own observations suggest that no appreciable decrease in morphine content need be expected even after several years' storage, provided the drug be well dried. We observed no' decrease whatever after storage for four years.
The few works containing data on the content or origin and distribution of the other alkaloids are predominantly qualitative studies. Some of the older publications must be viewed with caution, since the research in question was conducted, in part at leas t, with inadequate analytical aids. M. Kerbosch (21), who observed the formation of the alkaloids during growth with particular care, established the following order of importance: narcotine, codeine, morphine, papaverine, narceine, thebaine. Only narcotine and an "amorphous" alkaloid could be found in the seeds, which in part confirms and in part contradicts earlier findings (22-26). L. v. Itallie (27) summarized these data, together with the results of other studies conducted at the Pharmaceutical Institute at Leyden. Quantitative data on the formation of the by-alkaloids have been given by J. Tomko (loc. cit.), who, however, deals only with their total content. J. F. Reith, A. W. M. Indemans & W. R.. Becker (28) have also supplied 18 information regarding the total alkaloid content of ripe capsules and have compared various species. In an earlier work (29) these authors noted that the morphine content of the stem increased from the bottom to the top. According to C. Braga (30), there is neither decrease in the total alkaloid content nor displacement in the composition of the individual alkaloids when ripe poppy capsules age. K. K?ver & V. Cieleszky (31) ascertained the narcotine content of various varieties of poppy by means of a polarographic method. Quantitative data regarding the individual by-alkaloids in the ripe capsule are also given by W. Kussner (loc. cit.). These, however, must also be regarded as incomplete, since no account is taken of. narcotoline. Now, according to our investigations to date, the narcotoline content of ripe capsules in most cases exceeds the quantity of the other alkaloids several times over. We were also able to show with several varieties of poppy (32) that the narcotoline increases in quantity as the poppy ripens, and also in storage. G. Baumgarten (33) had already made similar observations regarding storage. These observations seem to show that the narcotoline, an appreciable quantity of which is present in the ripe capsule, originates from the partial demethylization of narcotine. This is also suggested by Kerbosch (loc. cit.), who ascertained by quantitative assay that large amounts of narcotine occur even in the young plant, and that this content increases with growth, but again decreases as the seed capsules ripen. Kerbosch found the highest content in the bud. H. Thoms (34) had already observed previously that before the plant blossomed there was appreciably more narcotine than morphine or codeine. Since according to Kerbosch codeine also occurs before morphine, it seems probable that in this case the o-demethyl alkaloids are the end-products of biosynthesis. This would admittedly conflict with the widely held view that the oxygen-methyl compounds are end-products, in which the methyl groups are fixed in such a way that they can no longer be mobilized by the organism (c.f.H.B. Schröter (35)).
The purpose of the procedures described below is to make it possible to supply quantitative findings on the formation of the by-alkaloids during growth and storage as well. The attempt to adapt the experiment for the investigation of opium also is prompted by the fact that the scientific investigation of the origin of opium plays an important part in the control of smuggling and illicit traffic (36). Conclusions regarding the origin of opium can be drawn from both quantitative and qualitative details of its alkaloid composition. In devising a method of this kind, special attention was paid to using only small quantities of drugs. In practice, this meant that only photometric or polarographic methods could be considered. As the alkaloids contained in an extract cannot be determined separately by photometric or polarographic means, a separation process had first to be devised; this we based partly on earlier studies (1-4).
The main difficulty was that, as against the classic methods of Anneler or Kljatschkina, precipitation reactions had to be discarded. The individual alkaloids could therefore be isolated only by extracting specimens of the drug with suitable solvents at various pH values. For this, it is essential that the individual alkaloids be completely separated in each case.
Unlike extraction or precipitation processes in macro-analysis, where small quantities of residual alkaloids can generally be neglected, micro-analysis may be seriously affected by such tests. In the method given below, the quantitative determination of the individual alkaloids is discussed and a method given for separating them quantitatively. In experiments conducted along these lines, consistent results were obtained throughout, and were confirmed in various ways by comparative evaluation and checked by paper-chromatographic methods, as described below.
QUANTITATIVE DETERMINATION OF INDIVIDUAL ALKALOIDS
Unfortunately, hope that Dyer & McBay's spectrophoto-metric process (loc. cit.), which enables alkaloids to be determined in the ultra-violet field, could be used for all alkaloids was not fulfilled, since there are still so many impurities present in the solutions of the individual alkaloids under investigation that any assessment, especially below a wavelength of 300 mµ, becomes impossible. Narcotine and papa-verine solutions are alone sufficiently purified by the various separation processes for their absorption not to be appreciably disturbed at 312 mµ.
Morphine
Given our aim of determining minute quantities of alkaloids, some gravimetric methods for determining morphine described in the literature may here be ignored. Similarly, of the numerous colour reactions of morphine, only a few are of use for quantitative determination, notably the formation of 2-nitrosomorphine, the diazo-reaction and the iodic acid reaction. H. Wieland & P. Kappelmeier (37) found that morphine in acid solution mixed with nitrite gives a yellow tint, which shades into orange-red when aqueous ammonia is added. This nitroso reaction was first suggested for the quantitative determination of morphine by R. Fabinyi (38). It was subsequently elaborated by F. A. Goin (39) into a quantitative method of determining the morphine in opium. The accuracy of the method, according to C. G. van Arkel (40), could be improved by using a Pulfrich photometer. Sarrat (41), E. F. Heeger & K. H. Bauer (42), and, more recently, D. C. M. Adamson & F. P. Handisyde (43), W. Poethke & E. Arnold (44), J. F. Reith, A. W. M. Indemans. & W. R. Becker (29), R. L; Stephens (45), E. Wegner (46), B. Acacic, D. Markovic & J. Petricic (47), A. Denoël (48), J. S. N. Cramer & J. G. Voermann (49), H. Baggesgaard-Rasmussen (50), A. B. Svendsen (51) and A. Bratina, M. Perpar & M. Tišler (52) also used this nitroso-morphine reaction for the quantitative determination of morphine, diacetylmorphine, and the morphine in opium and in parts of the poppy plant. The reaction has other uses too, since morphine can be made susceptible to polarographic determination - in opium and the poppy also - via the formation of 2-nitroso-morphine and its reduction (53-58). The diazo reaction is one often used for the quantitative colorimetric analysis of phenols. It is also the basis of the method of morphine assay proposed by L. Lautenschläger (59) and later, with modifications, by L. David (60) and by E. J. Ginsburg
|
Sample |
Morphine content |
Narcotoline |
Cotarnoline |
Total narcotoline |
Ratio of total narco- toline to morphine |
|---|---|---|---|---|---|
|
I. Kleinwanzleben |
0.23 | 0.022 |
<0.001 |
0.022 | 9.5 |
|
II. Hybrid |
0.36 | 0.017 |
<0.001 |
0.017 | 4.8 |
|
III Mahndorf |
0.29 | 0.019 |
<0.001 |
0.019 | 6.5 |
|
IV. Hybrid |
0.40 | 0.01 |
<0.001 |
0.01 | 2.5 |
|
V. Schlanstedt |
0.26 | 0.093 | 0.012 | 0.105 | 40.5 |
|
VI. Tourneur Freres, type B [a] |
0.68 | 0.048 | 0.014 | 0.062 | 9.1 |
|
VII. Blue poppy from Poland [b] |
0.35 | 0.16 | 0.02 | 0.18 | 51.3 |
|
VIII. Poppy from the East [a] |
0.37 | 0.197 | 0.013 | 0.21 | 56.7 |
|
IX. Native poppy 8 [a] |
0.61 | 0.136 |
0. 024 |
0.16 | 26.6 |
|
X. Unknown .. |
0.52 | 0.194 | 0.006 | 0.20 | 38.4 |
|
XL Russia 2 [a] |
0.48 | 0.13 | 0.005 | 0.135 | 28.2 |
|
XII. Tourneur Fr?s, type D[a] |
0.31 | 0.105 |
<0.001 |
0.105 | 34.0 |
|
XIII. Unknown |
0.51 | 0.154 | 0.016 | 0.17 | 33.3 |
|
XIV. Unknown |
0.33 | 0.075 | 0.008 | 0.083 | 25.2 |
|
XV. Unknown |
0.44 | 0.135 | 0.01 | 0.145 | 35.2 |
Origin: Schlanstedt variety.
bOrigin: Schlanstedt.
&N. J. Gawrilow (61). P. Balak & A. Jindra (62) also used this method recently. According to them, m-nitraniline and p-nitraniline are best suited for coupling. H. Auterhoff(63) investigated the quantitative ratio of the coupling elements and the optimum lye concentration for various substances, including morphine. The iodic acid reaction, first used for morphine determination by L. Georges & A. Gascard (64), was later successfully tried by A. Heiduschka & M. Faul (65) for the determination of morphine in poppy capsules. According to L. Szabolcz (66), solutions of morphine react with iodic acid by separating the iodine. When NaOH is
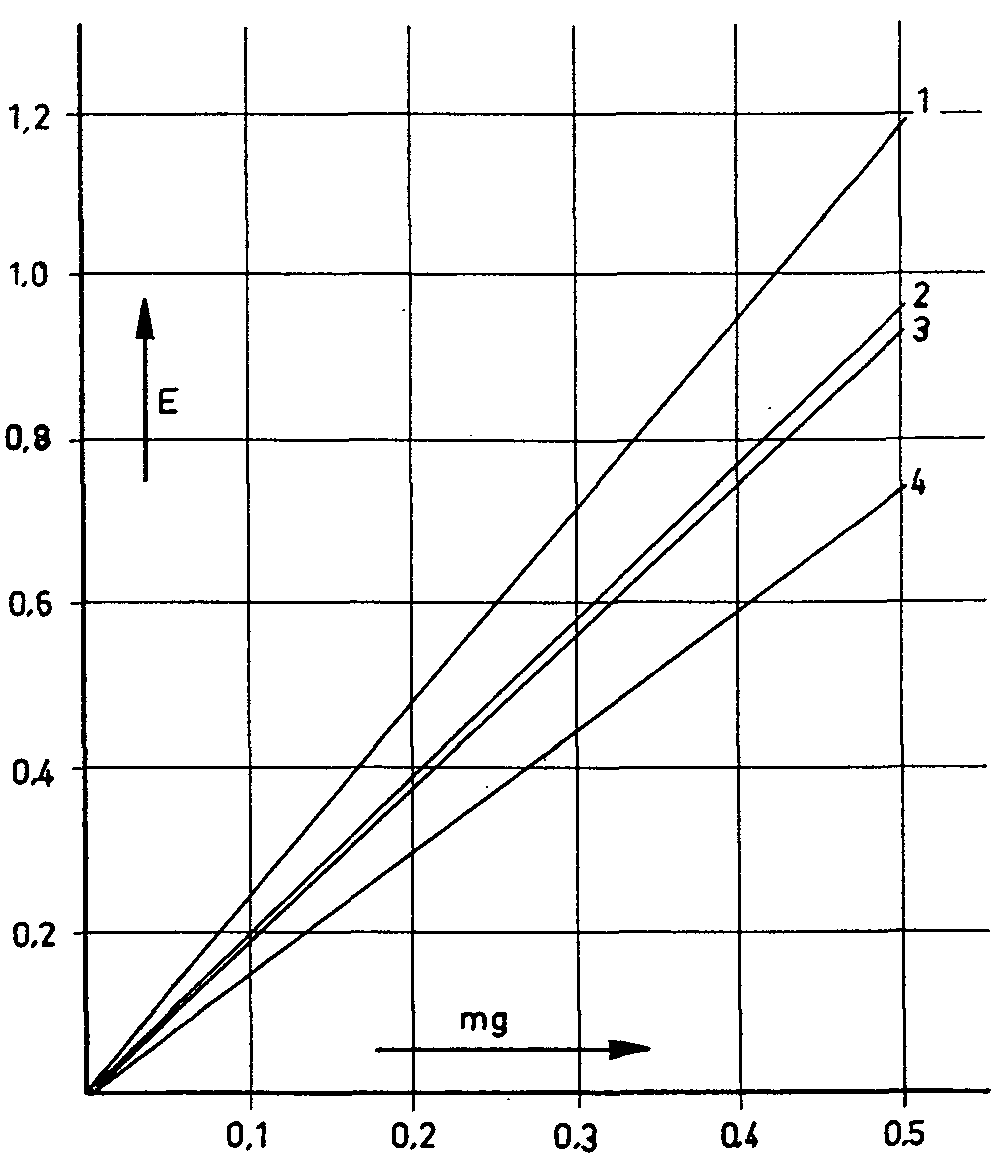
Figure 1
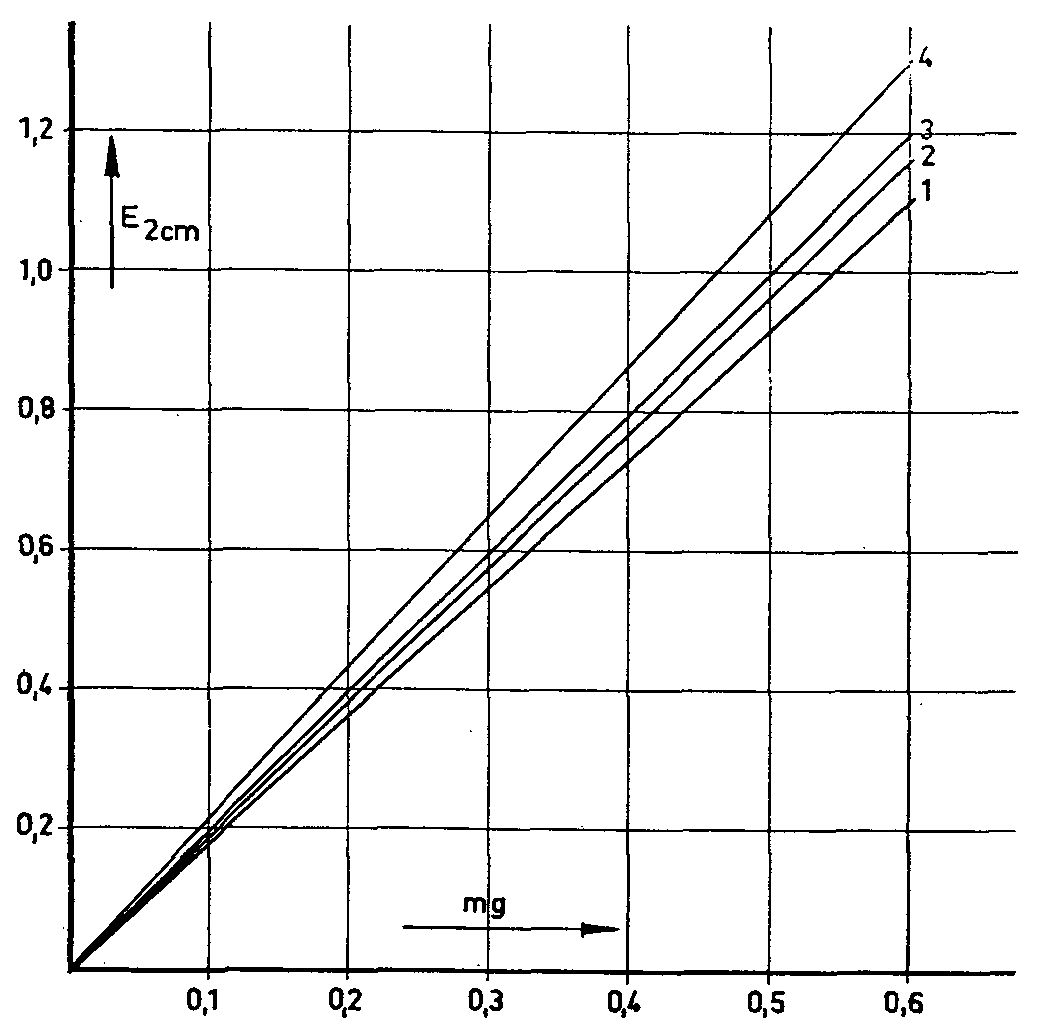
|
1. Morphine |
3. Morphine + 15 % narcotoline |
|
2. Morphine + 10% narcotoline |
4. Morphine + 30% narcotoline |
added, the solution turns orange-yellow. This colour, caused by the hydroxyl of the morphine, also lends itself to colori-metric determination. This reaction has recently been refined by A. Mariani, S. Guarino & O. Marelli (67) to an exact spectrophotometric method for the determination of the morphine in opium. C. F. Moorhoff (68) adopted this method for assay in poppy capsules. J. N. S. Cramer & J. G. Voermann (69) and R. R. A. Pride & E. S. Stern (70) made further modifications by using nickel salts instead of iron (III) chloride. E. Brochrnann-Hanssen (71) adopted the methods of the last-named authors in determining the morphine in opium after
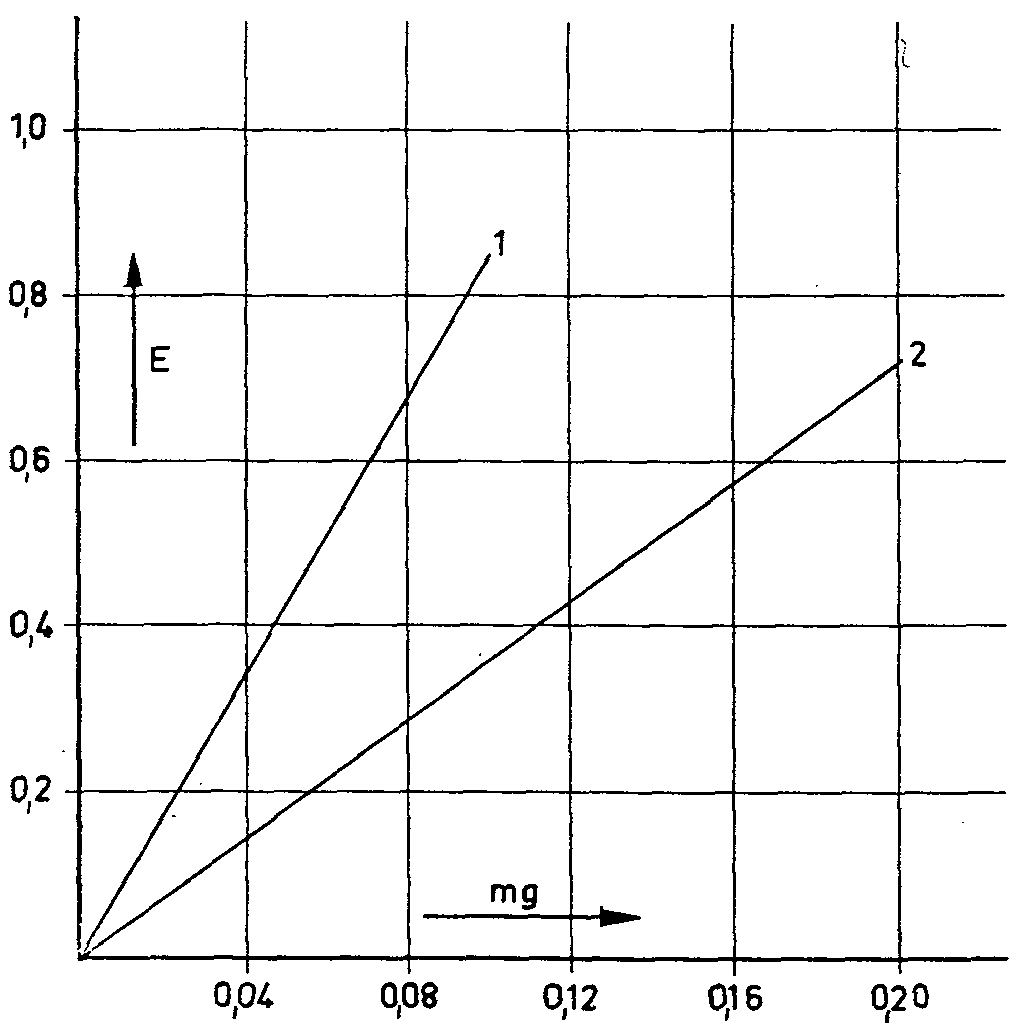
Figure 2
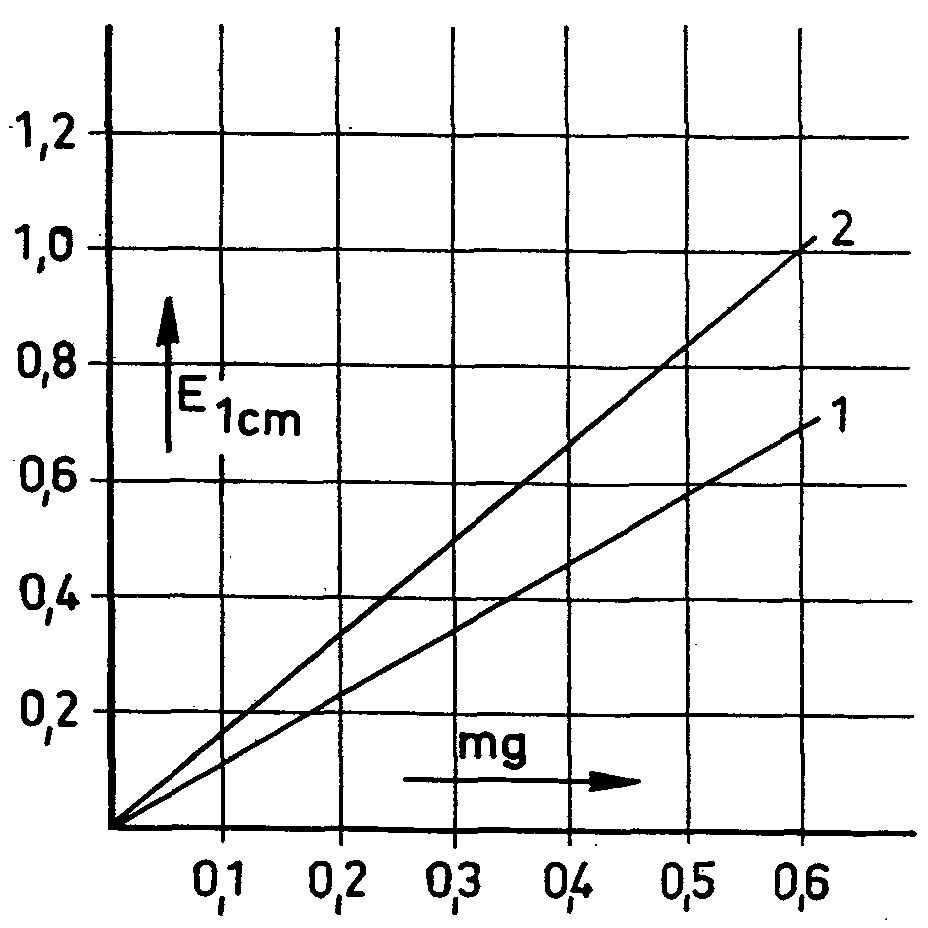
1. Morphine
2. Morphine + 10% natcotoline
previously separating the morphine with ion exchange resins. These reactions are based exclusively on the presence of a phenolic hydroxyl group in the morphine molecule. It is thus not surprising that various other phenol alkaloids present in the poppy plant and other phenols should give similar reactions; in general, therefore, the morphine must be exactly separated before the determination itself is undertaken. The separation of non-alkaloid phenols presents no difficulties in this connexion. Of the other poppy alkaloids with a phenolic hydroxyl group (laudanine, laudanidine, codamine and narcotoline), narcotoline alone occurs in the poppy in any considerable quantity, sometimes as much as 30% to 50% of the morphine level (72, cf. table I). It was therefore necessary to discover whether narcotoline reacted with the reagents mentioned by colouring. This it did in all cases. Subsequent checks of morphine and narcotoline mixtures showed that the morphine values were in each case affected by narcotoline to a greater or lesser extent (73) (figs. 1-4). Only appreciable amounts of narcotoline were taken into account in each case. These studies, which were also used for determining the morphine content of the poppy head, clearly showed not only that the presence of narcotoline sometimes gives incorrect results for morphine, but also that in some cases the requisite conditions simply do not exist, as in the determinanon of morphine in poppy capsules by Pride & Stem's method (loc. cit.). These authors invesngated the ioclic acid test described by-Cramer & Voermann (loc. cit.) to see whether it could not be adapted for the determination of morphine in opium and poppy capsules. They found that no poppy alkaloids give a colour reaction except pseudomorphine, which is sometimes detected in opium and in poppy capsules, and turns brown with iodic acid, and a deeper brown when an ammoniacal solution of nickel-chloride is added. In view of the difference in the extinction maxima (morphine; 670 mμ pseudo-morphine; 530 mμ), the authors used a correction factor to evaluate the morphine value arrived
Figure 3
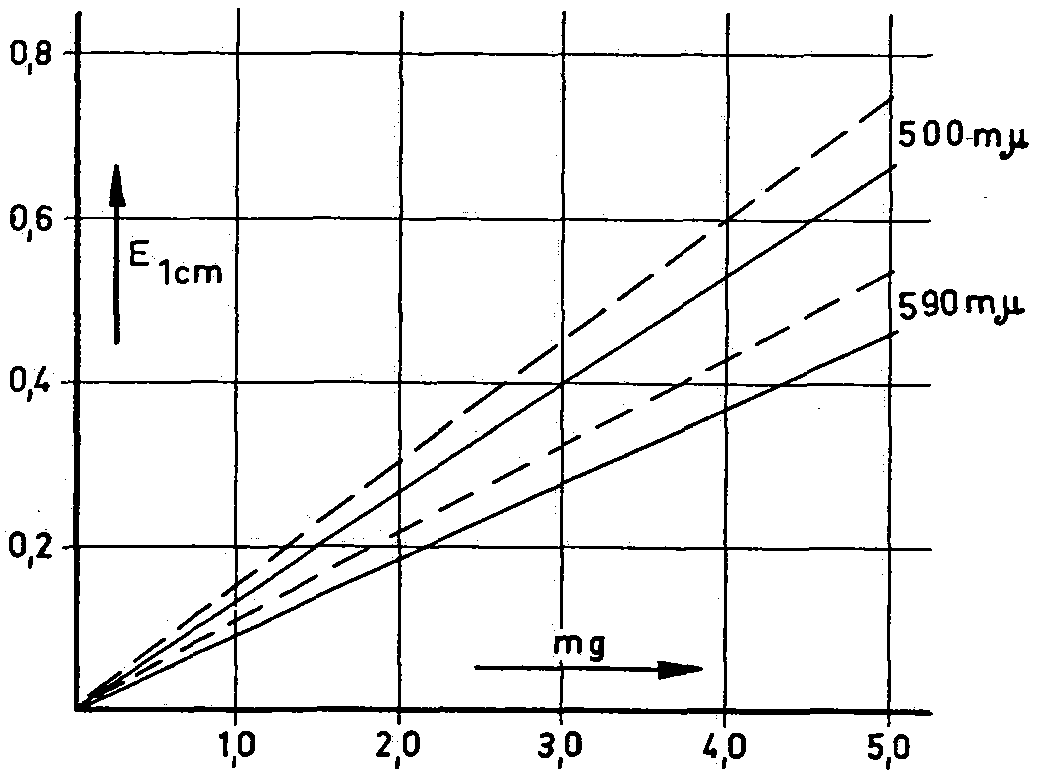
____________________Morphine
____ ____ ____ ____Morphine + 30% narcotiline
Figure 4

________________Morphine
___ ___ ___ ___Morphine + 30% narcotiline
at by computation and experiment, in order to eliminate error caused by pseudomorphine. In addition, the extinction was measured at both wavelengths. Narcotoline was disregarded. Our own experiments showed that narcotoline develops an olive-green colour [under experimental conditions], the intensity of which is overshadowed by the deep green of the morphine. Photometric measurements showed that narcotoline had practically, no effect on the morphine value at 660 mμ, but did show an effect at 530 mμ (Fig. 4), The relevant absorption curves (fig. 5) help to explain this. To extract the morphine from the poppy capsule powder
Figure 5
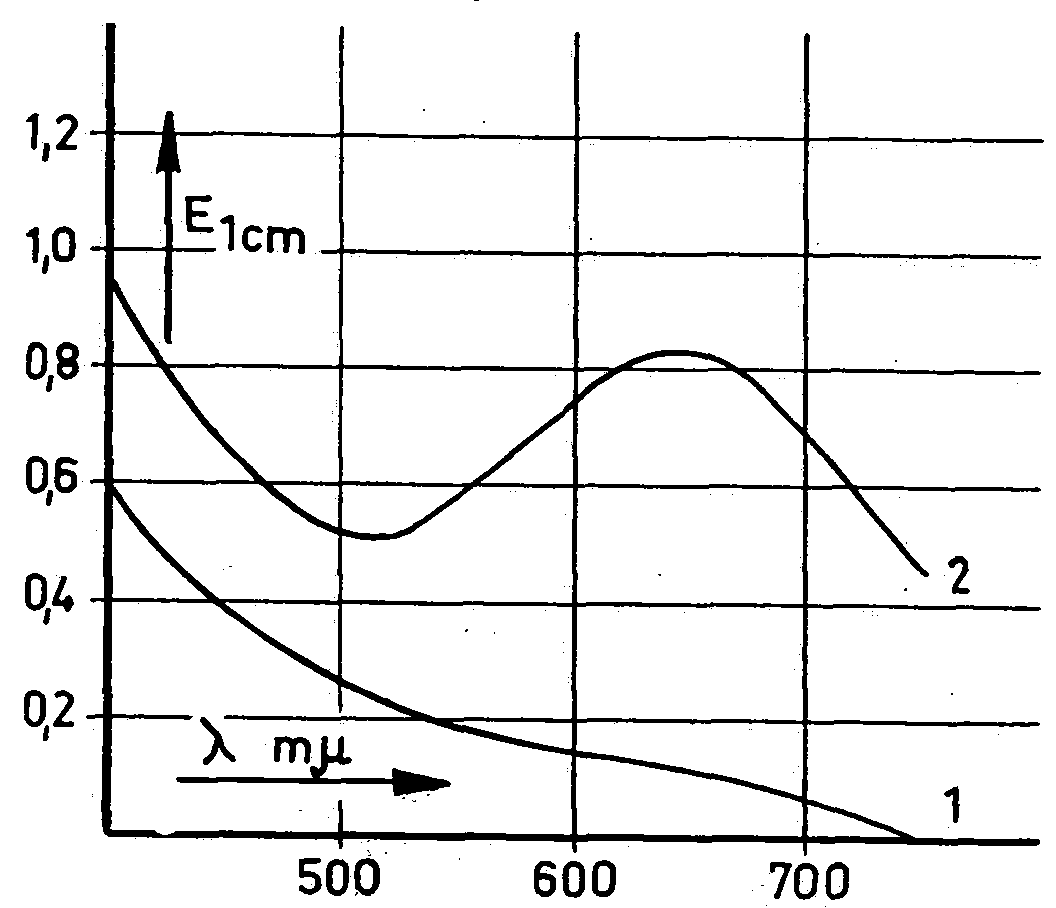
Figure 6

according to Pride & Stem's method, the former is first alkalized with a sodium-carbonate solution and shaken with a benzene-n-butanol mixture (1 + 1) for one hour, after which the solvent is drawn off by suction and the residue washed. The morphine is separated from the solution with sulphuric acid and determined by spectrophotornetric means in the sulphuric-acid solution without further purifying. The narcotoline thus passes almost completely into the sulphuric-acid solution. Although at 660 mμ (maximum absorption of morphine) it has no effect on the morphine level in the quantities in question, additional measurement at 530 mμ may give a false impression that pseudomorphine is present. The morphine figure is then "corrected" in error, and the value arrived at is a purely illusory one.
TABLE 2
Tests to compensate the effect of narcotoline on the nitrose reaction of morphine by heating the compensating solution
|
Extinction compared with | |||
|---|---|---|---|
|
Morphine (mg) |
Narcotoline (mg) |
Reagent solution |
Compensating solution heated in water-bath |
| 0.3 |
|
0.55 |
|
| 0.3 | 0.05 | 0.58 | 0.56 |
| 0.3 | 0.1 | 0.62 | 0.55 |
| 0.3 | 0.2 | 0.75 | 0.54 |
| 0.4 |
|
0.75 |
|
| 0.4 | 0.05 | 0.79 | 0.76 |
| 0.4 | 0.1 | 0.82 | 0.74 |
| 0.4 | 0.2 | 0.91 | 0.75 |
| 0.5 |
|
0.89 |
|
| 0.5 | 0.1 | 0.97 | 0.90 |
| 0.5 | 0.2 | 1.07 | 0.89 |
| 0.5 | 0.3 | 1.17 | 0.92 |
Narcotoline is also included during polarographic determination of morphine (57), but any erroneous results usually fall well within the margin of error (fig. 6; table 3). The nitroso reaction seemed the most suitable for the methods discussed here, its reliability and high degree of sensitivity having been demonstrated in numerous studies. Disturbances due to narcotoline can, moreover, be largely eliminated in fairly simple fashion, without complicated separation, since it can be dissolved in a slightly alkaline solution, as described later. The resulting cotarnoline turns a standard solution yellow in much the same way as the narcotoline in the test solution is tinted by nitrite and ammonia. A type of compensation thus takes place, as may be seen from table 2.
Special instructions for the selective determination of morphine in quantities of 0.1 to 0.3 g of the drug for the examination of poppy heads, or parts of poppy heads, are given in the experimental part, section IV (cf. also 57 and 74).
|
mg. alkaloid in 5 ml N/1 HCl | ||
|---|---|---|
|
Morphine |
Narcotoline |
mg morphine found |
| 0.2 | 0.1 | 0.22 |
| 0.2 | 0.05 | 0.2 |
| 0.35 | 0.1 | 0.365 |
| 0.25 | 0.12 | 0.265 |
| 0.18 | 0.06 | 0.185 |
| 0.3 | 0.09 | 0.32 |
Narcotoline
The phenol reactions to which reference has already been made (72) could also be evaluated quantitatively, but they are not suitable for determining the narcotoline unless it has previously been separated, owing to the usually excessive quantity of morphine present. The phenol reaction apart
Figure 7
from morphine can only be used after condensation with 4-amino-antipyrine in alkaline solution in the presence of an oxidizing agent (75). It is therefore worth noting that narco- toline can readily be separated in a slightly alkaline solution, especially if heated. This produces cotarnoline, an o-deso-methylcotarnine, and meconine :
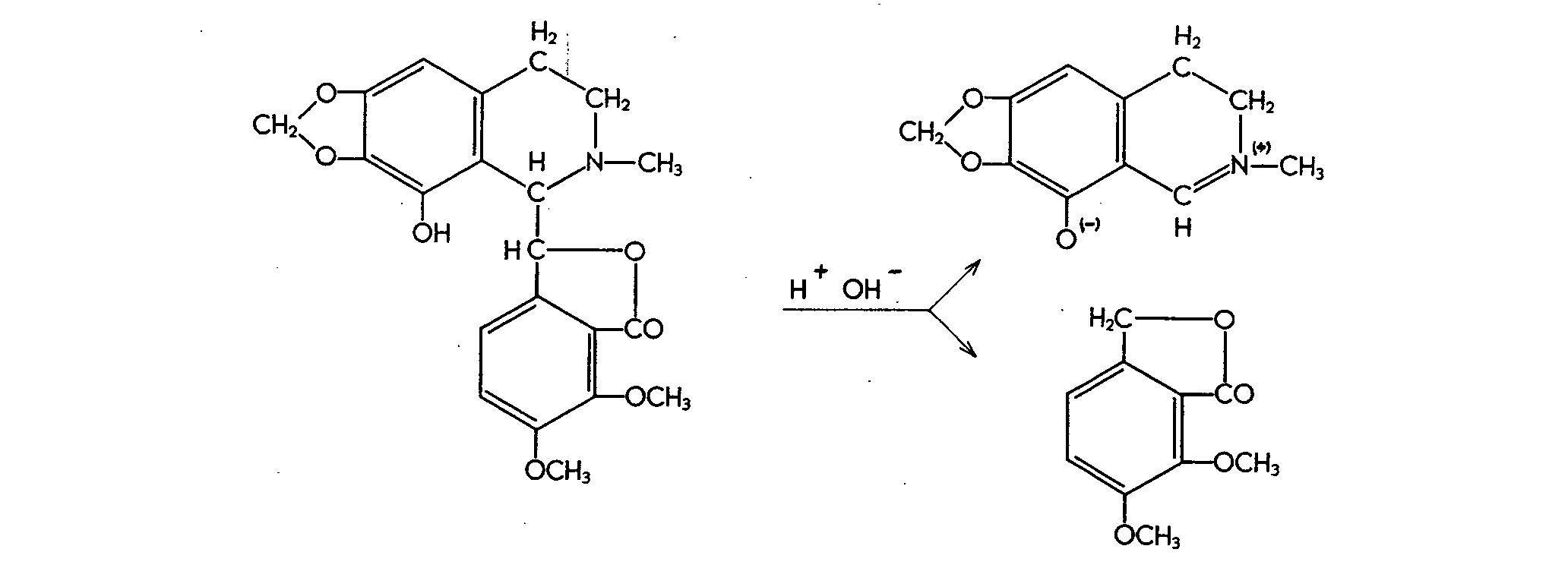
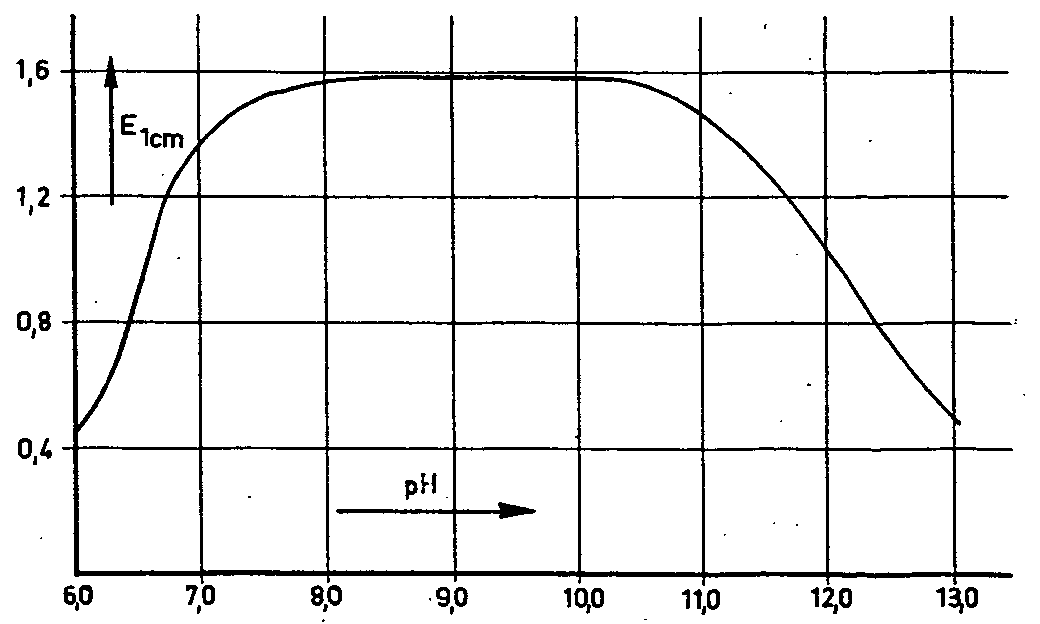
Ultraviolet and infra-red spectroscopic studies and polaro-graphic examination showed that both cotarnoline and its salts are in fact quaternary ammonium compounds, while pH titration showed the base itself to be an intrinsic salt. By contrast, cotarnine has an aldehyde structure, and assumes a quaternary ammonium pattern only in the salt form. It was found that the formation ofcotarnoline from narcotoline occurs quantitatively at pH = 8-10 (fig. 7). At pH values > 10 or < 8, conversion is not complete. The cotarnoline thus produced is bright yellow, and enables the narcotoline to be determined quantitatively from 0.05 mg upwards. Although narcotoline has recently been found in opium too (78), its determination in opium was disregarded owing to the low content (< 0.05%).
|
1. Extraction of alkaloids from opium with N/2 sulphuric acid; extraction from the poppy plant after alkalization of same with sodium-carbonate solution together with a mixture of methylene chloride, ether, and methanol in the ratio of 60 : 30 : 10. Following evaporation of the solvent mixture in a vacuum, the residue is absorbed with ether and shaken out with N/2 sulphuric acid. The sulphuric-acid solution is separated and (for poppy extracts only) mixed with trichloro-acetic acid; an aliquot portion is used for measuring the narcotoline; another is adjusted with aqueous ammonia to a pH of 7-8 and extracted with chloroform. |
|
|
|
2. Chloroform phase (Narcotine, papaverine, codeine, thebaine, narceine, morphine, and narcotoline in part only). Shake out with N/10 soda-lye |
3. Aqueous phaseMorphine, narcotoline, narceine) |
|
|
4. Chloroform phase |
5. Alkaline extract |
10. Acidify alkaline solution with concentrated |
|
(Narcotine, papaverine, codeine, thebaine) Shake out with 1 : 500 tartaric-acid solution |
(Morphine, narcotoline, narceine) Combine with aqueous phase (3) |
hydrochloric acid. Perforation with chloroform (opium only). |
|
6. Chloroform phase (Narcotine, papaverine) Evaporate chloroform, dissolve residue in alcohol, and heat with N/1 soda-lye over a water-bath; extract with benzene after cooling |
7. Acid extract (Codeine, thebaine) Alkalize. with soda-lye, shake out with chloroform. After compression of solvent and absorption of residue in 4 N/10 sulphuric acid, perform photometric measurement of codeine and thebaine |
11. Chloroform phase (Narceine) After evaporation of the chloroform, perform polarographic measurement of narceine |
|
8. Benzene extract (Papaverine) Extract with N/10 hydrochloric acid and measure papaverine by spectrophotumetry ar 312 mμ. |
9. Alkaline solution (Narcotine) Acidify with hydrochloric acid, heat in a water-bath; after cooling, mix with aqueous ammonia and extract with chloroform. After evaporation of solvent and absorption of residue in N/10 hydrochloric acid, measure narcotine by spectrophotometry at 312 mμ. |
12. Acid solution (Morphine, narcotoline) Alkalize with aqueous ammonia, shake out with chloroformisopropanol. After evaporation of solvent mixture, measure morphine by photometry |
Codeine and Thebaine
Quantitative photometric investigation of the thebaine and codeine content of poppy capsules has not yet been undertaken as far as we know. Since no simple method originally existed for separating codeine from thebaine, the first task was to assay the two alkaloids together. One advantage was that both alkaloids together can be determined with sufficient accuracy by means of the diazo reaction described by Wegner (79). Codeine can be oxidized to codeinone with potassium permanganate, and codeinone reacts with the diazo reagent to form a coloured compound. The excess of the oxidizing agent is eliminated with ferrous ammonium sulphate. Thebaine, as an enol ether of codeinone, can be transformed into the latter by acid hydrolysis, and undergoes the same colour reaction. According to Wegner, the determination of the thebaine with codeine is perfectly possible, but that of codeine with thebaine is not.
It was found that the speed of the oxidation of the codeine depends less on the time it takes the potassium permanganate to act than on the acidity of the solution. Wegner had already shown that the maximum extinction is reached after only 15 seconds. According to Wegner, a longer period (of oxidation) apparently leads to the formation of further oxidation products, which are likewise liable to undergo colouration in the presence of the diazo reagent. This is suggested, for example, by the differing tints (codeine, raspberry; thebaine, violet) and by the greater intensity in the case of thebaine. Unfortunately, it is not clear from Wegner's work what was the pH value of the codeine solution used for plotting the calibration curve. In the light of our findings, it must have been a very slightly acid solution; according to Wegner's rule, such a solution does in fact yield the highest extinctions. As the acidity of the solution is increased, oxidization of the codeine via codeinone to further oxidization products occurs appreciably faster (table 4); this is shown by the fact that
|
Codeine (mg) |
Acidity of solution (Sulphuric acid) |
Extinction |
|---|---|---|
| 0.01 |
N |
0.68 |
| 0.05 |
N |
0.65 |
| 0.1 |
N |
0.62 |
| 0.2 |
N |
0.53 |
| 0.3 |
N |
0.46 |
| 0.4 |
N |
0.35 |
| 0.5 |
N |
0.32 |
the longer the oxidization agent takes to act, the smaller the extinctions become, being practically nil after as little as one minute (table 5). On the other hand, it was also found that; under identical experimental conditions (length of oxidization = 15 seconds) solutions prepared with sulphuric acid yield extinctions considerably lower than those suggested by Wegner's curve, yet consistent with Lambert-Beer's law.
|
Extinction after oxidation | |||||
|---|---|---|---|---|---|
|
Acidity (Sulphuric acid) |
15 sec. |
30 sec. |
45 sec. |
60 sec. |
120 sec. |
|
N/100 |
0.68 | 0.67 | 0.65 | 0.65 | 0.61 |
|
4 N/10 |
0.35 | 0.22 | 0.07 | 0.03 | 0.01 |
Since oxidization of codeine in sulphuric-acid solution quickly leads (45-60 seconds) via codeinone to the formation of oxidization products which no longer colour in the presence of the diazo reagent, it seemed safe to assume that the 15-second period required for the oxidization of codeine might also be sufficient for oxidization of the relevant amounts of codeinone formed from thebaine to produce colourless solutions with the diazo reagent. This assumption proved correct when thebaine was hydrolized for 20 minutes and then treated in the same way as codeine (table 6). It is thus possible to
|
Thebaine (mg) |
Acidity of solution (Sulphuric acid) |
Extinction |
|---|---|---|
|
|
N/100 |
0.35 |
|
|
N/20 |
0.32 |
| 0.2 |
N/10 |
0.28 |
|
|
N/5 |
0.16 |
|
|
3N/10 |
0.09 |
|
|
4N/10 |
0.03 |
|
|
N/2 |
0.02 |
| 0.15 |
4N/10 |
0.01 |
| 0.10 |
4N/10 |
0.02 |
| 0.05 |
4N/10 |
- |
determine both alkaloids together. For determining the codeine one needs only to heat the sulphuric-acid solution containing both alkaloids before oxidization for some time in a boiling water-bath. The optimum acid concentration, on the basis of the findings given in tables 4 and 5, is 4N/10. With this degree of acidity, the codeinone already present at the beginning of oxidization is oxidized fast enough and the extinctions, even for minute quantities of codeine (0.04-0.2 mg) are still large enough to permit of sufficiently accurate measurement, as shown by table 7. The photometric determination of thebaine is somewhat less accurate than that of the other alkaloids (approximately ± 10 per cent). The reason for this is apparently that with hydrolysis not only codeinone, but also other products appear, as analysis of the hydrolosis products by paper chromatography showed. With increasing hydrolosis, for example, a second diazopositive compound occurs. To ensure sufficient accuracy, then, the time prescribed
|
Given |
Observed | ||
|---|---|---|---|
|
Codeine (mg) |
Thebaine (mg) |
Codeine (mg) |
Thebaine (mg) |
| 0.04 | 0.08 | 0.041 | 0.078 |
| 0.06 | 0.04 | 0.058 | 0.037 |
| 0.08 | 0.08 | 0.083 | 0.082 |
| 0.1 | 0.02 | 0.095 | 0.018 |
| 0.12 | 0.08 | 0.12 | 0.074 |
| 0.14 | 0.05 | 0.135 | 0.049 |
| 0.16 | 0.10 | 0.17 | 0.105 |
| 0.18 | 0.20 | 0.185 | 0.185 |
| 0.20 | 0.08 | 0.20 | 0.085 |
for hydrolosis must be exactly adhered to. Investigation of the hydrolosis products and the oxydization products of codeine under the conditions described has not yet been completed.
Narcotine and Papaverine
Certain methods for the quantitative photometric and polarographic assay of these alkaloids already exist (31, 79, 80, 81). Of these, only the polarographic method for determining the narcotine content of the poppy has apparently found practical application so far. The methods described by J.Holubek & J.Volke (79) and by K.K?ver & V.Cieleszky (loc. cit.) are based on splitting the narcotine by oxidation into cotarnine and opianic acid, which can be reduced by polarography.
For the photometric determination of the alkaloids referred to, E.Wegner has given a fluoroscopic photometric method for the determination of papaverine (81) and a method based on the oxidation of narcotine (79) with subsequent treatment of the reaction products with diazobenzene sulphonic acid. Since neither method proved entirely satisfactory in our investigations - least of all as applied to drug extracts - the ultraviolet spectrophotometric method seemed the most suitable. Both alkaloids have a maximum at 312 mμ. The method therefore presupposes the separation of the narcotine from the papaverine, which is easily done by splitting the lactone of the narcotine.
Narceine
No attempts have hitherto been made to determine the narceine contained in the poppy, the quantities observed in various paper-chromatographic investigations having been extremely small. The narceine content of opium must be determined polarographically, since the colour reactions described in the literature (82, 83) are so insensitive that the test sample would have to be about ten times as large before any useful results were obtained. In acid conductivity solutions, narceine effects a well-defined step in the bi-electronic reduction of the keto group conjugated with the double bond. Its half-stage potential is independent of the concentration of the alkaloid, but varies with the pH of the solution (84). In this connexion, it was important to note that meconic acid is also directly reducible by polarography (85). Furthermore, the half-stage potentials of these two compounds differ by only 0.2 volts, a fact which seriously interferes with the determination of the narceine through meconic acid - which is in any case present in opium in excessive amounts. It can, however, be shown that, under the conditions used for extracting narceine, the solution used for polarographic measurement is free from meconic acid.
THE SEPARATION PROCESS
The following points should be noted with regard to the method of separating the alkaloids given below.
The application of heat is avoided in extraction from the poppy plant. The usual methods of soxhlet extraction using methylene chloride (Kussner, Wegner, Poethke & Arnold, etc.), the preparation of hot extracts with benzene and alcohols (e.g., Baggesgaard-Rasmussen & Lanng), and the preparation of aqueous-acid extracts are all unsuitable, partly owing to the sensitivity of narcotolin (72) in slightly alkaline solution to heat, and partly owing to the formation of emulsions during subsequent isolation of the alkaloids.
For the extraction of 2.5-5 g of drug, 100 ml of the prescribed solvent mixture is normally sufficient. After additional extraction using a further 100 ml, paper chromatography revealed only small quantities of morphine, barely exceeding 1% of the total morphine content. In isolated cases, small quantities of cotarnoline were also identified in the paper chromatogram. A further extract prepared, using 100 ml of the solvent mixture, contained no further alkaloids. The instructions accordingly prescribe 150-200 ml of solvent mixture for purposes of extraction. Attempts to extract the alkaloids from the drug with various organic solvents or mixtures by shaking for several hours proved unsuccessful, since no quantitative determination of the alkaloid could be made, and because very large quantities of ballast material found their way into the organic solution when highly alcoholic solvent mixtures were used. For the investigation of opium a sulphuric acid extract is used.
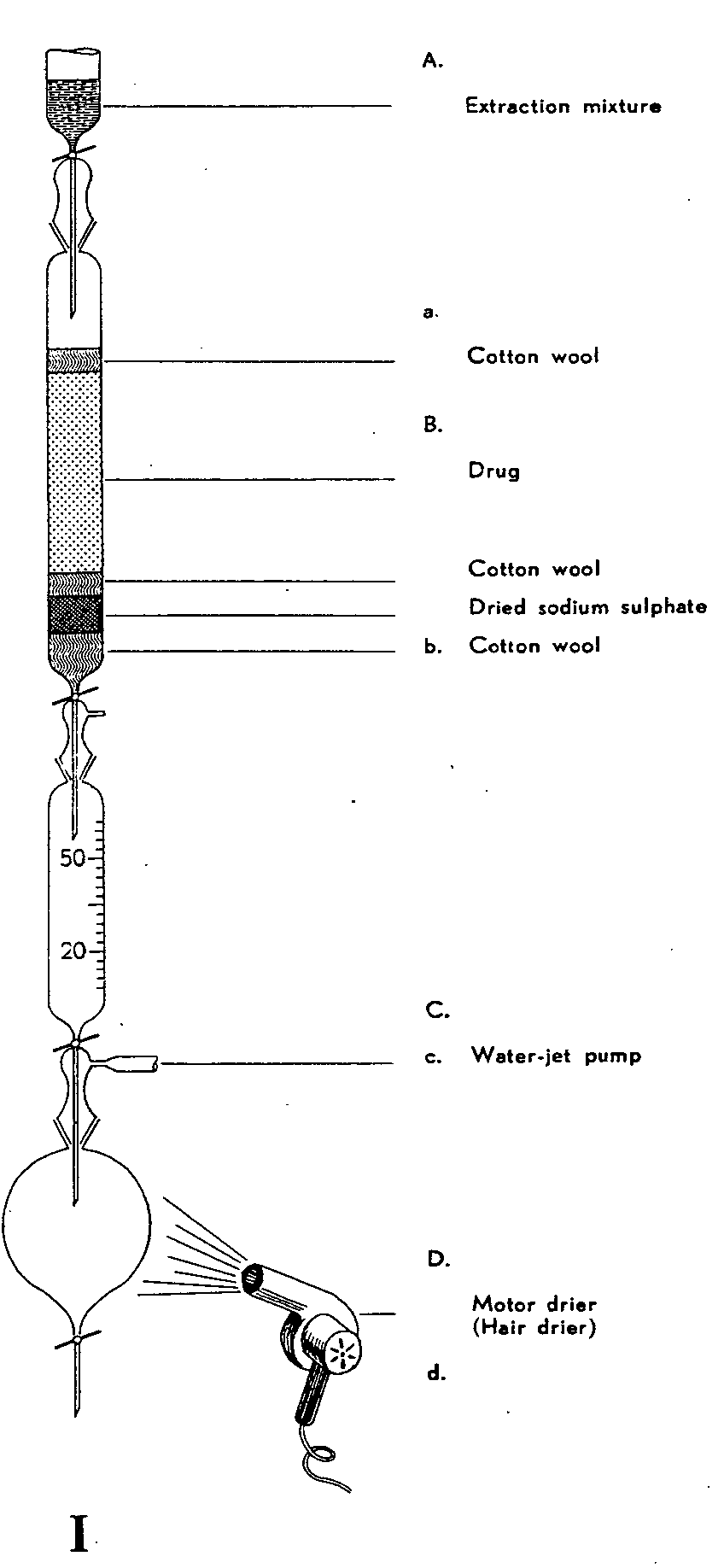
To save time, compression of the solution containing the alkaloid takes place in a vacuum at normal temperature during the actual extraction process (fig. 8, I).
The addition of trichloracetic acid to the acid-extract liquid helps to eliminate ballast materials and largely prevents emulsification during subsequent isolation of the individual alkaloids. Extraction with trichloracetic acid instead of sulphuric acid is inadmissible, since parts of the narcotine, papaverine and narcotoline may remain in the ether. When investigating opium, trichloracetic acid is not added, since it interferes with the polarographic determination of the narceine.
In order to simplify the method further, an attempt was made to extract narcotine, papaverine, thebaine, and codeine from the acid-extract solution after alkalization with soda-lye. However, paper chromatography showed that the alkaline liquid still contains narcotine and papaverine, presumably owing to the presence of dissolving agents. These
Figure 8
alkaloids were therefore first converted to the organic phase at pH 7.5-8.0 by means of chloroform. In this way, narcotine and papaverine can be determined quantitatively, as well as codeine and thebaine. However, since morphine, narceine, and narcotoline are also partly extracted at this pH, the latter have subsequently to be separated with soda-lye; during this process, the narcotine and papaverine are completely retained in the chloroform.
The narcotine and papaverine are separated from the codeine and thebaine by extracting the chloroform solution with a 0.2% tartaric-acid solution. Experiments with more concentrated solutions of tartaric acid showed that papaverine in particular passes partly into the acid phase. Conversely, attempts to separate the codeine and thebaine by using acid buffer solutions produced incomplete separation.
To separate narcotine from papaverine, the narcotine; in alkaline solution, is split with lactone. The papaverine can be extracted with benzene from the alkaline liquid, the narcotine of which is henceforth hydrosoluble.
The quantitative extraction of narceine proved extremely difficult. Neither frequent shaking out of hydrochloric-acid solutions with chloroform nor extraction with the same solvent at the iso-electric point met with success. The only method whereby narceine could be quantitatively converted from hydrochloric-acid solution into chloroform was by perforation, using a Schott perforator.
The alkaloids laudanine and laudanidine also had to be specially considered when investigating opium. Although they are phenol bases, they occur largely in the codeine-thebaine fraction, and are thus extractable with chloroform from sodium oxide alkaline solution, as previous reports have shown (86). This is of some consequence, since both alkalis colour in response to the diazo reagent. Despite the lesser colour intensity of these reaction products by comparison with that of codeine and thebaine, the danger of error could not be entirely excluded, particularly since paper chromatography of various kinds of opium showed the content to be not inconsiderable. When a sulphuric-acid solution, obtained in the course of analysis from commercial opium and containing laudanine and laudanidine as well as codeine and thebaine, was mixed with a diazo reagent and an alkaline lye, an orange tint with measurable absorptions, which could not originate either from thebaine or from codeine, did in fact appear. However, it was found that the orange tinting of the alkaloids with the diazo reagent absorbed practically no light at the wavelength of 570 mμ suited to the measurement of thebaine (the absorption maximum for laudanine and laudanidine is about 470 mμ), and that in the course of the operations required for performing the colour reaction for codeine (heating in sulphuric-acid solution, oxidation with potassium permanganate) laudanine and laudanidine also undergo changes and the resultant reaction products no longer give a diazo reaction.
With the narcotoline content, it is also possible to determine the content of cotarnoline (72, 76, 77). Generally speaking, however, the figure was included with that for narcotoline, since cotarnoline is not really a genuine alkaloid in its own right.
The separation process described was, as already indicated, checked for accuracy by paper chromatography, in accordance with a previously described method (87).
This was followed by the separation of the narcotine, papaverine, and narcotoline alkaloids, after splitting up the last-named into its cotarnoline component with ether saturated with aqueous ammonia. According to A. Bettschart (88), the separation of narcotine and papaverine can also be successfully performed on buffered paper (pH = 3.8) with water-saturated ether. Under these conditions, narcotoline can also be separated without previously decomposing the narcotine and papaverine (89). We accordingly adopted these variants in later experiments. Apart from traces of alkaloids not under discussion here, only the alkaloids expected were found in the individual fractions. The presence in the thebaine-codeine solution of laudanine and laudanidine, the separation of which by paper chromatography has not yet succeeded, has already been noted. Slight quantities of morphine which fell within the margin of error of the photometric method were found from time to time in the tartaric-acid solution of thebaine and codeine. In some cases this error can be eliminated by extracting the alkaline solution disintegrated with ammonium chloride and from which thebaine and codeine have previously been extracted with chloroform by means of a mixture of chloroform and isopropanol. This extract is added to the chloroform-isopropanol solution, which contains the largest quantity of morphine. The methods of photometric determination were also re-examined. The polarographic method of determining narcotine recently published by J. Holubek & J. Volke (70), for example, was found useful. Wegner's (loc. cit.) colour reactions for narcotine and his (loc. cit.) fluoro-photometric method for determining papaverine were also employed.
As already stated, neither method proved completely satisfactory. We were unable, for example, to reproduce the calibration curve for narcotine given by Wegner, obtaining instead a considerably flatter curve. Before being adopted by research laboratories not equipped with a universal spectrophotometer, these methods would require considerable revision and adaptation.
Figure 9
( - - - - - - - PH = 2.5/----------- pH = 7.2/ ................... PH = 12.2)
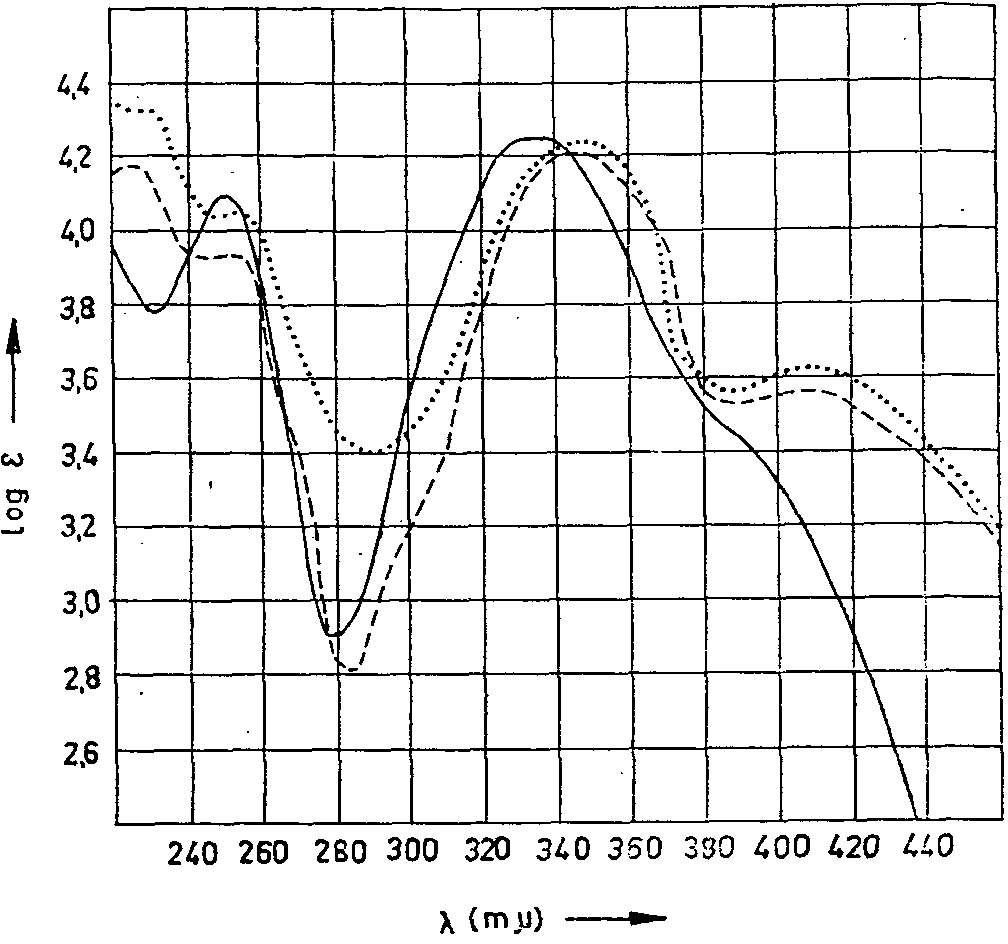
That the solutions employed for the spectrophotometric determination of narcotine and papaverine were sufficiently pure was proved by measuring the extinctions at various other wavelengths and by comparing them with pure solutions of the alkaloids of similar concentration. The values obtained for narcotoline were easily checked. The cotarnoline which occurs on decomposition of narcotoline, besides the maximum used for determination at 410 mμ also has maxima between 338 and 348 mμ (fig. 9) with considerably larger extinctions, both in acid and in alkaline solution. We attached special importance to comparative measurements at these wavelengths, since at 410 mμ only the yellow tint is measured; results might thus be affected by contamination caused by extractives. Finally, polarographic investigations (cotarnoline) can be reduced by polarograph (77) and also closely agree with photometric findings. The findings for morphine were also checked by polarograph (57). For codeine and thebaine, in particular, for which no comparative checks were possible and all other alkaloids, too, paper chromatography also demonstrated the correctness, by and large, of the values found. Lastly, some of the alkaloids were also determined by a recently developed method for quantitative paper chromatography by photographic means (90),
It should be added that, in the investigation of leaves or other parts of the poppy plant with a very low alkaloid content disturbances may be caused by emulsion formation owing to the high chlorophyll content or because of the larger weighed portion of drug sometimes necessary in certain cases. In such cases, it may be necessary to separate the aqueous from the organic phase by means of centrifugation.
Experimental part1. Analysis of Parts of the Poppy
5 g of poppy capsules or 5.0 g of other parts of the poppy (in each case finely ground) are well saturated with 2 or 4 ml respectively of sodium carbonate solution (7.5 g anhydrous sodium carbonate, 92.5 g water) and left to stand for 30 minutes pressed against the sides of the mortar. At the end of this time, the moist powder is collected and poured into extraction tube B on to approximately 5 cm of dried sodium sulphate, as shown in fig. 8, I. From the reserve container A, a mixture of 60 parts by volume of methylene chloride, 30 parts ether, and 10 parts methanol is fed through tap b until the extract begins to drip into collecting vessel C. Tap b is then closed, to ensure better saturation of the drug with the solvent. At the end of 15 minutes, tab b is again opened and extraction continued without interruption, the flow through tap b beingadjusted to approximately 80 to 100 drops per minute. The solution collected in container C is allowed to pass from time to time via tap c into the separating funnel D; the solvent in D is then brought to evaporation by suction with a water-jet pump. To avoid excessive icing of the separating funnel, it is advisable to apply a mechanical drier (e.g., a hair-drier) from the side. Extraction takes about 1? to 2 hours. The residue is absorbed with 10 to 15 ml ether and is twice shaken out for 2 minutes with 10 ml of N/2 sulphuric acid. The acid extracts are collected in an Erlenmeyer flask and mixed with 1.0 g trichloro-acetic acid; the whole is well shaken, allowed to stand for a few minutes, and passed through a dry circular filter of 8 cm diameter into a 25-ml measuring flask. The Erlenmeyer flask and filter are twice rinsed with 2 ml of water and the contents of the measuring flask then diluted with water up to 25 ml (= solution A). 10 ml of this solution are used for further determination of narcotoline; the remaining 15 ml are rinsed with a little water, passed into a separating funnel, and adjusted to pH 7.5 to 8.0 by adding drops of 10% aqueous ammonia. The pH value of the solution is checked by means of a suitable indicator paper. When the solution passess from acid to alkaline values, its colour darkens, thus greatly facilitating adjustment of the pH. The solution, now slightly alkaline, is next shaken three times with 20 ml of chloroform, each time for 2 minutes, after which the combined chloroform solutions are extracted with 10 and 5 ml of N/10 soda-lye.[1] The alkaline solutions are added to the extracted fluid adjusted to pH 7.5 to 8.0 (= solution B), this solution is mixed with 2 ml of hydrochloric acid DAB.6 (DAB = German Dispensatory), poured into a 50-ml measuring flask, and mixed with 5 ml of 10% aqueous ammonia. The flask is filled up to the mark, after which the morphine is extracted from 10 ml (more in some cases) of this liquid with 20 and 2 ? 10 ml of chloroformisopropanol (3 + 1 parts by volume). The combined organic solutions are then compressed in the water-bath, the residue is absorbed with 5 ml of chloroform, and the latter shaken out with 20 ml of N/10 hydrochloric acid (= solution C).
The codeine and thebaine are separated from the chloroform solution extracted with soda-lye by shaking them with 10 and 2 ? 5 ml of 0.2% tartaric-acid solution. The combined tartaric acid solutions are mixed with 1 ml of 15% sodalye and twice extracted with 20 ml of chloroform. The organic solvent is evaporated over the water-bath and the residue absorbed with 10 ml of 4N/10 sulphuric acid, the whole being slightly warmed (= solution D). The remaining chloroform solution, containing narcotine and papaverine, is compressed, the residue mixed with 2 ml of alcohol and 5 ml of N/1 soda lye, and heated for 5 minutes in a boiling water-bath. After cooling, the mixture is extracted three times with 5 ml of benzene; the benzene solutions are then shaken with 5 ml of N/10 sodalye once more, and the alkaline solution of the liquid containing lactone-separated narcotine is added. To isolate the papaverine, the benzene solution is extracted with 10 ml of N/10 hydrochloric acid (= solution E). To regenerate the narcotine, the solution containing it is mixed with 2 ml of 25% hydrochloric acid and heated for 30 minutes in a boiling water-bath. After cooling, aqueous ammonia is added until an alkaline reaction sets in and extraction performed twice with 20 ml chloroform; the solvent is
|
mg |
mg |
Poppy capsules % |
Opium % | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Alkaloid |
Given |
Observed |
Given |
Observed |
Observed |
Added |
Total |
Observed total |
Observed |
Added |
Total |
Observed total |
|
Morphine |
4.0 | 3.9 | 3.0 | 3.1 | 0.58 | 0.2 | 0.78 | 0.76 | 10.6 | 3.0 | 13.6 | 13.9 |
|
Codeine |
0.2 | 0.21 | 0.5 | 0.46 | 0.027 | 0.02 | 0.047 | 0.044 | 0.82 | 0.5 | 1.32 | 1.27 |
|
Thebaine |
0.15 | 0.17 | 0.20 | 0.19 | 0.008 | 0.02 | 0.028 | 0.030 | 0.54 | 0.5 | 1.04 | 0.98 |
|
Narceine |
0.2 | 0.18 | 0.10 | 0.11 |
- |
- |
- |
- |
0.24 | 0.2 | 0.44 | 0.47 |
|
Papaverine |
0.5 | 0.48 | 1.0 | 1.05 | 0.015 | 0.10 | 0.115 | 0.107 | 1.2 | 1.0 | 2.2 | 2.08 |
|
Narcotine |
0.5 | 0.50 | 2.0 | 1.95 | 0.047 | 0.10 | 0.147 | 0.140 | 3.3 | 3.0 | 6.3 | 6.5 |
|
Narcotoline |
1.0 | 0.94 | 0.5 | 0.53 | 0.125 | 0.05 | 0.175 | 0.170 |
- |
- |
- |
- |
|
Cotarnoline (included as narcotoline) |
- |
- |
- |
- |
0.030 |
- |
0.030 | 0.030 |
- |
- |
- |
- |
then compressed over the water-bath and the residue absorbed with 10 ml of N/10 hydrochloric acid. After filtration, this solution (= solution F) is used for the spectrophoto-metric determination of narcotine.
2. Analysis of' Opium
1 g of opium is accurately weighed, carefully crushed in a mortar with a few drops of N/2 sulphuric acid, and further diluted up to a total of 10 ml. The solution is poured into a measuring flask of 25 ml capacity and the mortar rinsed with 2 x 5 ml of N/2 sulphuric acid. The combined solutions are then diluted with water up to 25 ml. 15 ml of this filtrate are used for the separation processes described above. The narcotoline is determined at this stage. For morphine and narceine, special procedures apply (cf. II, lb and II, 6).
For measuring the extinctions, when analysing opium, it may be necessary to use thinner vessels than those described. If necessary, the solutions must also be diluted (narcotine, codeine).
1. Morphine
(a) Determination in Parts of the Poppy
5 ml of filtered solution C are poured into a test-tube with a ground stopper, mixed with 2 ml of a 1% sodium-nitrite solution, and immediately shaken for 15 seconds. Exactly 15 minutes after the addition of the nitrite, the combination is mixed with 3 ml of 10% aqueous ammonia, after careful rotation with 2.5 ml of water. Five minutes later, with the aid of a suitable photometer,[2] the colour intensity is measured at 470 mμ (2-cm vessel) against a standard solution[3] prepared in the following way :
5 ml of solution C mixed with 3 ml of 10% aqueous ammonia and 2.5 ml of water, heated for five minutes in a boiling water-bath, and subsequently cooled.
(b) Determination in Opium
The acid liquid remaining in the Schott perforator after separation of the narceine is carefully poured, together with the chloroform also remaining there, into a separating funnel; the chloroform is separated and the acid solution poured into a measuring flask of 100 ml capacity. The perforator and the separating funnel are well rinsed with small amounts of water. 10 ml of 10% aqueous ammonia are added, and water is added to the solution in the flask up to the mark. The morphine is extracted from 20 ml of this solution with 20 and 2 x 10 ml of chloroform-isopropanol (3 + 1 parts by volume), the combined organic solutions are compressed in the water-bath, the residue absorbed with 5 ml of chloroform, and the latter shaken with 20 ml of N/10 sulphuric acid. After filtration, 5 ml of this solution are used for the determination of morphine, as explained under II, 1a above.
2. Codeine
3 ml of solution D are heated in a boiling water-bath for 20 minutes in a test-tube in which a smaller tube filled with water is suspended to avoid evaporation of water. After cooling, the solution is mixed with 0.5 ml of a 0.3% solution of potassium permanganate; the whole is immediately shaken, and oxidation is interrupted after exactly 15 seconds by adding 0.5 ml of a freshly prepared 10% solution of ammonium ferrosulphate. After 5 minutes, 2 ml of 15% soda-lye are added, after which the mixture is allowed to stand for one hour. It is then separated by filtration from the sediment, and 3 ml of the filtrate are mixed with 5 ml of a mixture of equal parts of diazo reagent I and II (DAB 6). The intensity of the ensuing raspberry colour is immediately measured against a similarly treated standard preparation at 500 mμ (2-cm vessel).
3. Thebaine
3 ml of solution D are heated for 20 minutes, as for codeine, and the resultant product mixed after cooling in cold water with 2 ml of 15% soda-lye and subsequently with 3 ml of a mixture of equal parts of diazo reagent I and II (DAB 6). The intensity of the ensuing violet shade is measured at 570 mμ (1-cm vessel).
|
Type |
Morphine |
Codeine |
Thebaine |
Narceine |
Papaverine |
Narcotine |
Narcotoline |
Cotarnoline (calculated as narcotoline) |
|---|---|---|---|---|---|---|---|---|
|
Poppy I |
0.41 | 0.027 |
- |
- |
0.008 | 0.06 | 0.075 | 0.015 |
|
Poppy II |
0.58 | 0.027 | 0.008 |
- |
0.015 | 0.047 | 0.125 | 0.030 |
|
Poppy III |
0.67 | 0.032 | 0.01 |
- |
0.018 | 0.022 | 0.050 | 0.024 |
|
Poppy IV |
0.71 | 0.043 | 0.015 |
- |
0.04 | 0.035 |
0. 11 |
0.035 |
|
Poppy V |
0.53 | 0.035 |
- |
- |
0.009 | 0.050 | 0.09 | 0.01 |
|
Opium I |
10.6 | 0.82 | 0.54 | 0.24 | 1.2 | 3.3 |
- |
- |
|
Opium II |
11.7 | 1.20 | 0.95 | 0.32 | 0.8 | 5.3 |
- |
- |
|
Opium III |
9.9 | 0.7 | 0.42 |
Less than 0.1 |
1.2 | 2.8 |
- |
- |
4. Narcotine, papaverine
The tinctures of solutions E and F are measured at 312 mμ (1- or 2-cm vessel). (1- and 2-cm vessels respectively).
5. Narcotoline
10 ml of solution A are brought with aqueous ammonia as near as possible to a pH = 7 and subsequently extracted with 20 and 2 x 10 ml of a mixture of 4 parts by vol. of chloroform and 1 part amyl alcohol. The organic solution is then shaken with 10 ml of N/2 sulphuric acid, and 4 ml of the acid solution are mixed with 1 ml of 10% aqueous ammonia; a further 4 ml are mixed with 1 ml of water (= standard solution) and the intensity of the yellow shade produced in the ammoniacal solution is measured at 410 mμ (1-cm vessel).[4] By this means one can determine the amount of cotarnoline present in the drug; this is best included with the narcotoline (cf. also under 9). After measurement, the ammoniacal solution is poured into a test-tube and heated for 5 minutes in a boiling water-bath. It is then cooled and the tincture is measured once more as before. This represents the amount of narcotoline, including cotarnoline, present in the drug.
6. Narceine
To determine the narceine content of opium, solution B (cf I, 1) is mixed with 3 ml of concentrated hydrochloric acid and perforated with chloroform in a Schott perforator
Figure 10
|
1 papaverine (312 mμ 1-cm vessel) |
3 narcotine (312 mμ 1-cm vessel) |
|
2 morphine (470 mμ 2-cm vessel) |
4 narcotoline (410 mμ 1-cm vessel) |
for about two hours. The solvent is then distilled, 5 ml benzene and 3 ml of N/10 soda-lye are added to the residue, the flask is closed, and shaken hard. 2 ml of 30% acetic acid are next added and the mixture is poured into a separating funnel. The acid component is separated, filtered, and used for the polarographic measurement of narceine. Two drops of gelatine solution are added, after which nitrogen is passed through the solution for 5 minutes. Measurements [5] are taken from - 0.8 volts at 20°C (E = 1/5); evaluation is performed in the usual manner, the narceine values being read off a calibration curve.
The amounts of alkaloid corresponding to the extinctions observed can be either read off calibration curves or calculated from the following ratios, obtained from the calibration factors given by the curves in figs. 10 and 11:
Morphine % = E 1 cm . 3.54 a
Codeine % = E 1 cm . 0.317 a
Thebaine % = E 1 cm . 0.069 a
Narcotine % = E 1 cm . 0.175 a
Papaverine % = E 1 cm . 0.0695 a
Narcotoline % = E 1 cm . 0.41 a
E 1 cm = extinction observed (calculated for a 1-cm vessel) a = weighed portion in g.
1. First Method (photometric)
1 to 0.2 g of poppy-capsule powder dried at 75°C. are heated in a centrifuge glass with 5 ml of N/10 acetic acid for ten minutes over a boiling water-bath, the whole being shaken from time to time. After cooling in cold water, 0.5 g of lead acetate is added, the centrifuge glass closed with a rubber stopper, and shaken hard for one minute. 0.5 g of dried sodium sulphate is added, the mixture again shaken hard, water added up to the level of about 10 ml, and the whole centrifuged for about three minutes at 3,000 to 5,000 revolutions per minute. The solution obtained in this way, which should be only slightly cloudy, is transferred to a separating funnel, while the residue is stirred and centrifuged twice more with 2.5 ml of N/10 acetic acid. The pH value of the combined solutions - which should be approximately 5.5 to 6.0 - is next determined with a suitable indicator paper.
Figure 11
| 1 |
thebaine |
(570 mμ ; 1-cm vessel) |
| 2 |
codeine |
(500 mμ ; 2-cm vessel) |
Where necessary, a small amount of sodium acetate may be added. The extract is next shaken twice, each time for two minutes, with 20 ml of chloroform, the chloroform solution is separated, and the solution to be analysed is mixed with 2 ml of 5% soda-lye and 0.3 g of ammonium chloride. The solution (pH approximately = 9) is extracted with 2 x 20 and 1 x 10 ml of chloroform-isopropanol (3 + 1 parts by vol.), each time for two minutes. After distillation of the solvents over a water-bath, the residue is absorbed with 5 ml of chloroform. From this, the morphine is drawn off with 10 ml of N/10 hydrochloric acid. After separation of the layers, the main portion of the hydrochloric solution is carefully poured off through a filter. 5 ml of the (clear, colourless) solution are placed in a test-tube fitted with a glass stopper, mixed with 2 ml of a 1% sodium-nitrite solution, and immediately shaken hard for 15 seconds. Exactly 15 minutes after addition of the nitrite, the sediment is mixed with 3 ml of 10% aqueous ammonia, and, after careful rotation, with 2.5 ml of water. After 5 more minutes, the extinction is measured against a standard solution consisting of 5 ml of N/10 hydrochloric acid and the above-mentioned amounts of nitrite solution, aqueous ammonia, and water (470 mμ, 2-cm vessels). The amount of morphine corresponding to the extinction observed may be read off a calibration curve. The morphine content of the capsules may be calculated from the formula
|
Morphine %= |
a . 2 . 100, |
where |
|
|
b |
|
a = the amount of morphine read off the calibration curve;
b = the measured portion.
2. Second Method (photometric)
2 g of finely powdered poppy capsules dried at 75°C are powerfully crushed in a mortar with 0.5 to 1 ml of water (in the case of coarser powder, 0.5 g of quartz sand should be added), and 15 minutes later partially ground fine with approximately 5.0 g aluminium oxide "acid". This gives a dry powder, which must be free from any coarse capsule parts. The powder is placed in a glass tube approximately 1.5 cm in diameter and 15 cm in length, the narrow end of which is closed with cotton wool and covered with approximately 3 g of aluminium oxide "acid ". This is next washed out with water, which is fed drop by drop from a separating funnel placed on the tube in such a way that, at a speed of 20 drops per minute (regulated by means of a water-jet pump), a layer of water 1 cm deep constantly covers the aluminium oxide.[6] Some 20 ml of the extract are absorbed, rinsed with 2 ml of water, and transferred to a separating funnel. 1 ml of aqueous ammonia (DAB 6) and 0.2 g of ammonium chloride are added, after which extraction is performed with 2 x 20 ml and 1 ? 10 ml of chloroform-isopropanol (3 + 1 parts by volume), for two minutes in each case. The combined solutions are heated over the water-bath while the solvent is distilled. The residue is absorbed with 5 ml of chloroform, after which extraction is performed with 15 ml of N/10 hydrochloric acid. 5 ml of the hydrochloric solution are then treated as explained under 1 above, for purposes of photometric measurement. A further 5 ml of this solution are mixed with 4.5 ml of water and 3 ml of aqueous ammonia and heated for 5 minutes in a boiling water-bath. This solution here serves both as a standard solution and also to compensate the narcotoline contained in the solution. The amount of morphine is calculated from the formula
|
Morphine % = |
a . 3 . 100, |
where |
|
|
b |
|
a = the amount of morphine read off the calibration curve;
b = the measured portion.
3. Third Method (polarographic)
0.3 g of finely powdered poppy capsules dried at 75°C are prepared as under IV, 2, above, and extracted, the amount of aluminium oxide being increased in this case to 7-8 g. Approximately 20 ml of the extract are absorbed and, after rinsing with 3.5 ml of hydrochloric acid (DAB 6), placed in a measuring flask of 25-ml capacity. Water is then added up to the mark. To 5 ml of this extract (= 0.06 g poppy-capsule powder), 2 ml of N/1 sodium-nitrite solution are added and, exactly 5 minutes later, 3 ml of 20% potash-lye and 7 drops of gelatine solution. Prior to polarographic measurement, nitrogen is passed through the solution for 5 minutes. During polarographic measurement, bottom mercury is used as the supply electrode from - 0.2 volts at 20°C. By using anelectrolysis vessel as the polarographic cell, it is possible to perform analysis in a vacuum. The polarograms are calculated by extending the lines of the fundamental current and the limit current to right and left respectively. In addition, an auxiliary line is drawn as a tangent through the centre of the rising part of the curve, and the difference in height between the points of intersection of these tangents is measured against the extended lines of the fundamental and limit currents. Finally, the morphine values are read off a calibration curve. The entire process - including waiting time, but excluding photographic work - takes about 45 minutes.
A method is described for the quantitative assay of the principal alkaloids in the poppy and in opium by micro-measurement, based on the isolation and separation of the alkaloids by extraction at various pH-values and subsequent photometric or (for narceine) polarographic measurement. A brief survey of the main works already published dealing with the occurrence (= geographical distribution), formation, and content of the poppy alkaloids is also given.
1In Poland, for example, narcotine, codeine, and thebaine (6, 7) are obtained from ripe poppy heads as well as morphine; efforts will obviously be made to produce a variety exhibiting the highest possible content of by-alkaloids.
aOrigin: Schlanstedt variety.
bOrigin: Schlanstedt.
1Sometimes the soda-lye is not cleanly separated after the second extraction. In such cases, the main portion of the chloroform should be removed, a few ml of ether added, and the whole shaken once more. This ensures complete separation. The soda-lye is then removed and the ether-chloroform mixture added to the chloroform previously removed.
2For all analyses, the universal spectral photometer of VEB Carl Zeiss, Jena, was used.
3This solution serves to compensate for any narcotoline that may be present (39).
4On addition of the aqueous ammonia or after heating, the solution usually becomes somewhat cloudy; filtration through a small circular filter is therefore sometimes necessary.
5In these studies, a micropolarograph, Heyrovsky system, with photographic registration of the voltage curve, was used.
6Apparatus 8,II has proved successful for this extraction process.
ANNELER, E., Festschrift Emil Barell, Basel 1936, p. 344.
002KLJATSCHKINA, B., Arch. Pharmaz. Ber. dtsch. Pharmaz. Ges. 272 , 558 (1934).
003ADAMSON, D. C., HANDISYDE, F. P. & H. W. HODGSON, Quart. J. Pharm. Pharmacol. 20 , 218 (1947).
004DYER, M. S. & A. J. McBAY, J. Amer. Pharm. Ass., Sci. Edit. XLIV, 156 (1955).
005KUSSNER, W., Mercks Jahresbericht 1940, 29.
006LUDWICKI, H. & P. GÓRECKI, Acta Polon. Pharmac. 13, 447 (1956); ref. Pharmaz. Zhalle 96, 221 (1957).
007BINIECKI, S. & L. RYLSKI, Ann. Pharm. franç. 14, 208 (1956).
008MALIN, A., Ber. dtsch. Pharmaz. Ges. 17, 60 (1907).
009FUCHS, L., Pharm. Mh. 13, 223 (1932); ref. C. 1933, I, 1476.
010GUILLAUME, A. & J. FAURE, Ann. Pharm. franç. 4, 160 (1946).
011MÜLLER, A., Arch. Pharmaz. 252 , 280 (1914).
012BAGGESGAARD-RASMUSSEN, H. & O. LANNG, Dansk Tidsskr. Farm. 22 , 201 (1948); ref. C.A. 1948, 9079.
013WEGNER, E., Pharmazie 6, 420 (1951).
014TOMKO, J., Farmacia (Bratislava) 24, 327 (1953).
015KUCERA, M., Ceskoslov. Farmac. 4, 308 (1955).
016POETHKE, W. & E. ARNOLD, Pharmazie 6 , 406 (1951).
017JESPERSEN, J. C., Dansk. Tidsskr. Farm. 10, 16 (1936); ref. C. 1936, I, 2389.
018v. KABAY, I., Ber. ung. Pharm. Ges. 12, 387 (1936); ref. C. 1937, I, 1729.
019WUEST, H. M. & A. J. FREY, Festschrift Emil Barell, Basel, 1936, p. 556.
020WEGNER, E., Pharmazie 8, 839 (1953).
021KERBOSCH, M. G. J. M., Arch. Pharmaz. 248, 536 (1910).
022ACCARIE, Jahersber. der Chem. IV, 250 (1835); cit. from Kerbosch (21).
023SACC, Jahresber. Fortschr. der Pharm. 64, (1848); cit. from Kerbosch (21).
024MEUREIN, Journ. de Pharm. 23, 339 (1853); cit. from Kerbosch (21).
025DIETERICH, Helfenberg. Annalen 1884, 75; cit. from Kerbosch (21).
026CLAUTRIAU, Rec. Inst. bot. Brux. II, 266; cit. from Kerbosch (21).
027v. ITALLIE, L., Ann. pharm, franç. 4, 156 (1946).
028REITH, J. F., INDEMANS, A. W. M. & W. R. BECKER, Pharmac. Weekbl. 83, 449 (1948); ref. C. A. 1948, 8422.
029Id., Pharmac. Weekbl. 82, 581 (1947); ref. Pharmaz. Zhalle 88, 52 (1949).
030BRAGA, C., Arch. Pharmacol. sperim. 53, 255 (1932).
031K?VER, K. & v. CIELESZKY, Acta pharmac. hung. 26, 12 (1956).
032PFEIFER, S., unpublished.
033BAUMGARTEN, G., Pharmazie 9, 97 (1954).
034THOMS, H., Arb. Pharm. Inst. Berlin 6, 215 (1909).
035SCHRÖTER, H. B., Pharmazie 10, 141 (1955).
036Report on the 12th session of the Commission on Narcotic Drugs of the Economic and Social Council of the United Nations (29.-31.5.1957) Dtsch. Apotheker Ztg. 97, 597 (1957).
037WIELAND, H. & P. KAPPELMEIER, Liebigs Ann. Chem. 382, 306 (1911).
038FABINYI, R., Chemiker Ztg. 35, 1099 (1911).
039GOIN, F . A., An. Farmac. Bioquim. 5, 93 (1934); ref. Chem. Zbl. 1935 I, 3569.
040VAN ARKEL, C. G., Pharmac. Weekbl. 72 , 366 (1935); ref. Pharmaz. Zhalle 76, 410 (1935).
041SARRAT, Quart. J. Pharmac. Pharmacol. 10 , 466 (1937); ref. C. 1938 I, 4501.
042HEEGER, E. F., & K. H. BAUER, Landwirtsch. Jb. 90 , 397 (1940).
043ADAMSON, D. C. M., & F. P. HANDISYDE, Quart. J. Pharmac. Pharmacol. 19 , 350; cit. from Wegner (46).
044POETHKE, W. & E. ARNOLD, Pharmaz. Zhalle 88 , 1 (1949).
045STEPHENS, R. L., J. Pharmac. Pharmacol. 3 , 815 (1951).
046WEGNER, E., Pharmazie 6 , 55 (1951).
047ACACIC, B., D. MARKOVIC & J. PETRICIC, Acta Pharm. Jug. 1, 3 (1951).
048DENOËL A., J. Pharm. Belgique 1 , 241 (1946).
049CRAMER, J. N. S. & J. C. VOERMANN, Acta Pharm. Int. 1 , 219 (1953).
050BAGGESGAARD-RASMUSSEN, A ., Ann. pharm. franç. 10 , 693 (1952).
051SVENDSEN, A. B., Dansk Tidsskr. Farmac. 22 , 131 (1955).
052BRATINA, A., M. PERPAR & M. TIŠLER, Mikrochim. Acta 1957 , 167.
053BAGGESGAARD-RASMUSSEN, A., C. HAHN & K. ILVER, Dansk Tidskr. Farma. 19 , 41, 71 (1945); cf. also 50 .
054NOSEK, J. & O. KRASTANOVA, Cas. ces. L?c?rn. 63 , 49 (1950).
055MATSUMOTO, K., J. Pharm. Soc. (Japan) 72, 1393, 1396, 1398 (1952); ref. C.A. 47 , 2430 (1953).
056HOLUBEK, J., Pharmaz. Zhalle 94 , 347 (1955); 95, 435 (1956).
057PFEIFER, S. & W. KELLER, Pharmaz. Zhalle 95 , 189 (1956).
058SCHULEK, E. & K. BURGER, Acta Pharmac. hung. 25 , 49, 193. (1955).
059LAUTENSCHLÄGER, L., Arch. Pharmaz. 257 , 13 (1919).
060DAVID, L., Pharmaz. Ztg. 76 , 706 (1931).
061GINSBURG, E. J. & N. J. GAWRILOW, J. anal. Chem . (russ.) 1, 282 (1946); ref. Chem. Zbl. 1947 I, 741.
062BALAK, F. & A. JINDRA, Casopis ceského lekárnictva , vedecka priloha 63, 125 (1950); ref. C. 1956 , 8163.
063H. AUTERHOFF, Tagungsbericht Arch. Pharmaz. Ber. dtsch. pharmaz. Ges. 290/62 , 132 (1957).
064GEORGES, L. & A. GASCARD, Journ. de Pharmacie et de Chimie 25 , 513 (1906) cit. from Wegner (46).
065HEIDUSCHKA, A., & M. FAUL, Arch. Pharmaz. 255 , 172 (1917).
066SCABOLCS, L., Magyar Kémiai Folyóirat 59 , 67 (1953); ref. C. 1955 , 10083.
067MARIANI, A., S. GUARINO & O. MARELLI, Rend. Ist. super. Sanité 14 , 733 (1951).
068MOORHOFF, C. F., Pharmac. Weekbl. 87 , 593 (1952); ref. Z. analyt. Chem. 140 , 230 (1953).
069CRAMER, J. N. S. & J. G. VOERMANN, Pharmac. Weekbl. 84 , 129 (1949).
070PRIDE, R. R. A. & E. S. STERN, J. Pharmacy Pharmacol. 6 , 590 (1954).
071BROCKMANN-HANSSEN, E., Medd. norsk farmac. Selsk. 17 , 76 (1955); ref. C. 1957 , 5647,
072PFEIFER, S. & F. WEISS, Pharmazie 10 , 658 (1955).
000BULLETIN ON NARCOTICS l JULY-SEPTEMBER 1958 33
073PFEIFER, S. & F. WEISS, Pharmazie 10, 701 (1955).
074PFEIFER, S., Pharmazie 11, 387 (1956).
075PFEIFER, S. (unpublished).
076PFEIFER, S. & F. WEISS, Arch. Pharmaz. Ber. dtsch. Pharmaz. Ges. 289/61, 24 (1956).
077PFEIFER, S., Arch. Pharmaz. Ber. dtsch. Pharmaz. Ges. 290/62, 261 (1957).
078PFEIFER, S., Arch. Pharmaz. Ber. dtsch. Pharmaz. Ges. 290/62, 209 (1957).
079WEGNER, E., Pharmazie 5, 33 (1950).
080HOLUBEK, J. & J. VOLKE, Pharmazie 11, 577 (1956).
081WEGNER, E., Pharmazie 5, 445 (1950).
082WEISSBERG, S. M., FIALKOW, J. A. & E. G. CHRISMAN, Pharmazie (UdSSR) 10, 26 (1947); ref. C. 1947, II, 1390.
083WITTE, A. H., Pharmac. Weekbl. 91, 588 (1956); ref. C. 1957, 203.
084KIRKPATRIK, H. F. W., Quart. J. Pharm. Pharmacol. 18, 338 (1949).
085HOBZA, J. & F. ŠANTAVÁ, Casopis ceského Lecárnictva 62, 86 (1949).
086The story of "porphyroxine-Meconidine" Bulletin on Narcotics, 1952, Vol. 4, No. 1.
087PFEIFER, S., Scientia pharmac. 24, 84 (1956).
088BETTSCHART, A., Thesis, Zürich, 1954.
089MIRAM, R. & S. PFEIFER (unpublished).
090MIRAM, R. & S. PFEIFER, Pharmaz. Zhalle 96, 457 (1957).

