Abstract
Introduction
Experimental
Results and discussion
Author: Carlton E. TURNER,, Cheng Y. MA, Mahmoud A. ELSOHLY,
Pages: 71 to 76
Creation Date: 1979/01/01
A method for the determination of cocaine content in coca leaves ( Erythroxylum coca) has been developed. The procedure involves refluxing the powdered leaves in 95 per cent ethanol for 15 minutes, followed by acid-base partitioning with chloroform and a GLC assay. The recovery of cocaine was quantitative. This procedure was applied to determine cocaine content in three samples of Erythroxylum coca Lam. collected from different geographic locations in Peru. Using androst-4-ene-3, 17-dione as the internal standard the calibration curve was linear over a factor of 0.5 to 10 fold cocaine concentration relative to internal standard. The slope ( b) was 0.733, the coefficient of determination ( r 2) was 1.00 and the average precision was 3.9 per cent.
It has been reported that the alkaloid content of South American coca leaves is about 0.5 to 1.5 per cent with cocaine representing 75 per cent of the total alkaloid content of Bolivian coca leaves. The total alkaloid content of Java coca leaves bas been reported as somewhat higher (1.0 to 2.5 per cent), but with cocaine representing only 50 per cent of the total alkaloids [ 1] . Farnsworth, et al. [ 2] were able to detect only trace amounts of cocaine in herbarium specimens in eight species of the genus Erythroxylum. The same group has also observed that the content of cocaine decreases significantly during storage. Mass fragmentography was used by Holmstedt et al. [ 3] to determine the cocaine content in 13 South American coca species. Cocaine concentration in the cultivated species ranged from 0.13 to 0.68 per cent and no cocaine was detected in the seeds.
In the past two decades, various methods for analysing pure cocaine and its metabolites have been developed and critically reviewed [ 4] . Since most procedures utilizing GLC techniques were directed toward the quantitation of cocaine in either biological fluids or pharmaceutical products [ 5] - [ 12] , 1 it seems necessary to develop an analytical method which is feasible for routine determination of cocaine and other alkaloids in coca leaves.
Apparatus: Gas chromatographic determinations were carried out on a Beckman GC-65 gas chromatograph equipped with dual flame ionization detectors.
1. These references are used as examples and are by no means inclusive.
The silanized glass column (2.4 m x 2 mm i.d.) was packed with 6 per cent OV-1 on 100/200 mesh Chromosorb W, AW, DMCS. The column, injector and detector temperatures were 220 °C, 295 °C, and 300 °C, respectively. Nitrogen was used as the carrier gas with a flow rate of 25 ml/min. The air and hydrogen were set at 300 ml/min and 50 ml/min, respectively.
Chemicals: Cocaine-free base was isolated from Erythroxylum coca Lam. (Tingo Maria) (m.p. 96-97 °C) and has been spectroscopically and GC-MS confirmed. Internal standard, androst-4-ene-3,17-dione (m.p. 170°-172 °C, S.S.T. Corporation New York, N.Y.) was used without further purification. Ninety-five per cent and absolute ethanol were of standard reagent grade. Chloroform was freshly distilled in glass.
Plant material: Coca leaves were collected from three locations in Peru (Cuzco, Trujillo and Tingo Maria) by Senior Ing. J. Alejandro Spirgatia Costa, De Empresso De La Coca, Lima, Peru. The plant material was identified by Professor M.W. Quimby, Department of Pharmacognosy, School of Pharmacy, University of Mississippi. Voucher specimens were stored in the Herbarium, Department of Pharmacognosy, School of Pharmacy, University of Mississippi.
Extraction procedure: Air-dried coca leaves were forced through a 9 mesh sieve and 1.00 g of the resulting finely manicured material was refluxed in 40 ml 95 per cent ethanol for 15 minutes. The mixture was filtered through Whatman No. 4 filter paper, and filtrate was evaporated under vacuum in a rotary evaporator at 40-50 °C until dry. The residue was then quantitatively transferred into a separatory funnel with the aid of 20 ml of chloroform and partitioned with 10 ml citric acid, 1.5 per cent (W/V). The aqueous layer was washed with an additional 20 ml of chloroform and adjusted to pH 8.2 with solid NaHCO3. Cocaine was then extracted twice from the aqueous layer using 20 ml of chloroform. The chloroform layer was dried over anhydrous sodium sulphate and evaporated to dryness under vacuum. The residue was then sonicated with 1.00 ml internal standard stock solution (10 mg/ml in ethanol) and 3 ml ethanol. An aliquot 0.1-1.0 μl) of the final solution was subjected to GLC analyses.
Calibration curve: To 100 μl of 1.0 mg/ml ethanolic solution of cocaine, a suitable amount of internal standard was added so that the ratio of their concentrations ranged from 0.1 to 2.0. The calibration curve was constructed by plotting the ratio of area under cocaine and internal standard peaks versus the ratio of their concentrations (figure 1). The response factor (or the slope b) and coefficient of determination ( r 2) were determined from the least square line. The reproducibility of the response factor was checked routinely. The quantity of cocaine was calculated as follows:
Wt (cocaine) = Wt (I.S.) X 1/b X Area (cocaine)/Area (I.S.)
The percentage of cocaine in leaf samples reported throughout the study was the mean of triplicate determinations.
Recovery: The over-all recovery of cocaine was carried out by analysing 1 g of leaf samples to which 5 mg amounts of cocaine standard were added and thereby the combined concentration of cocaine was measured. The original cocaine content in the plant material was determined in a separate extract without the addition of the standard. For the study of recovery from aqueous solution, 5 mg of cocaine were added to 10 ml of 1.5 per cent citric acid and extracted according to the procedure described above.

Calibration curve - A standard curve for cocaine was constructed by plotting the area ratio of cocaine and internal standard versus the ratio of their concentration at 1 mg/ml of cocaine concentration (figure 1). The response factor (or the slope b) and the coefficient of determination ( r 2) determined from the least square line were 0.733 and 1.00 respectively. For routine determination of cocaine concentration from coca leaves, the response factor was checked routinely with freshly prepared cocaine standard solution.
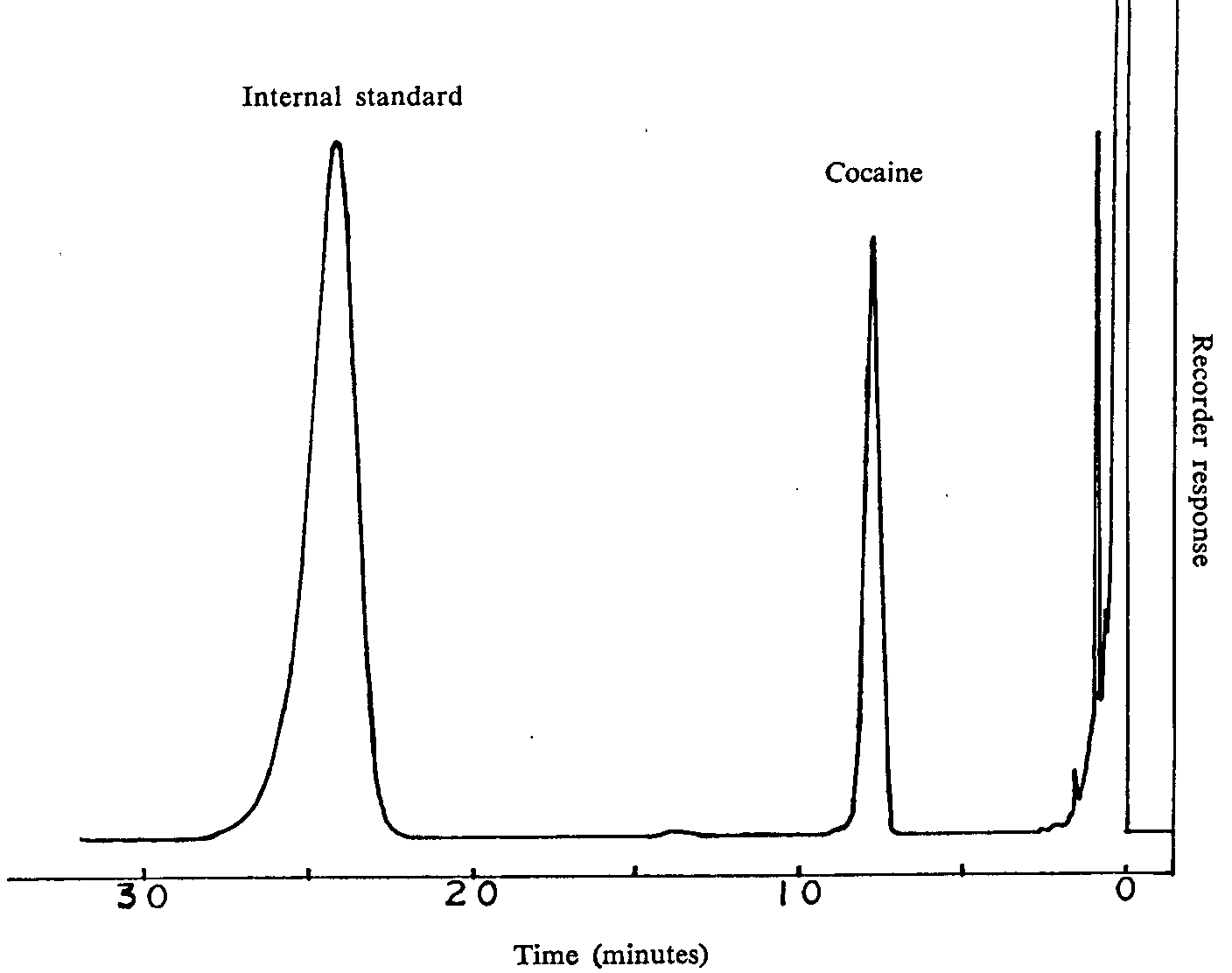
Extraction procedure - Table 1 gives a summary of the concentrations of cocaine in coca leaves obtained from one hour, single-step extraction using various solvent systems. As indicated, the method involving refluxing with ethanol for 15 minutes offered the most efficient result. The only disadvantage associated with the single-step extraction procedure is the high residue content (about 38 per cent) which interferes with the GLC determination of cocaine (relative retention time=0.32, with retention time of 24.3 minutes for internal standard). Removal of potential interferences of acidic and neutral components was achieved by acid and chloroform partitioning steps. Back extraction of cocaine from basified aqueous fraction with chloroform yields relatively low residue content (1.5 per cent). A typical chromatogram is shown in figure 2. Efficiency of extraction was evaluated by refluxing the plant material with ethanol for 15, 30, 45 and 60 minutes. In all cases, comparable amounts of cocaine were recovered. However, long refluxing times resulted in lower precision (table 2) and thus 15 minutes was chosen as the ideal time.
|
Solvent system a |
Cocaine b (percentages) |
Coefficient of variation (percentages) |
|---|---|---|
|
Ethanol
|
0.13 ± 0.00
|
0.0 |
|
Ethanol reflux b
|
0.60 ± 0.03
|
5.0 |
|
Methanol
|
0.28 ± 0.08
|
29.0 |
|
Methanol/NH
4OH (99.5:0.5)
|
0.32 ± 0.08
|
25.0 |
|
Methanol/NH
4OH (96:4)
|
0.37 ± 0.07
|
19.0 |
|
Methanol/Diethylamine (3:1)
|
0.43 ± 0.04
|
9.3 |
|
Ethanol/Diethylamine (3:1)
|
0.42 ± 0.04
|
9.5 |
aMean of triplicate determinations.
bSolvent extraction was carried out for one hour in all cases except for ethanol refluxing, which was carried out for 15 minutes.
Recovery of cocaine - Cocaine has a pKa of 8.6 and so is best extracted into organic solvents at alkaline pH, preferably below 10 [ 5] . Both rapid extraction and minimal contact with acid and base result in the most efficient extraction and are essential to prevent hydrolysis (4). Initially, various combinations of acid and base were used to evaluate the recovery of cocaine standard from aqueous solutions. We found that the combination of 2 per cent HCI (pH = 0) and concentrated NH 4OH (pH=9-9.5) yielded a 93 per cent recovery, a result which compares well with the recovery rate reported elsewhere [ 4] (93.3 per cent at pH=9.5). However, quantitative recovery was achieved by using 1.5 per cent citric acid (pH=2) and saturated NaHCO 3 (pH=8.2). Using this combination of acid and base in the partitioning step and analysing the cocaine spiked plant material, quantitative overall recovery was determined. This indicated that no hydrolysis of cocaine takes place during the 15 minute reflux. As a result of optimization of solvent system used and refluxing time, the entire extraction procedure can be carried out in less than 45 minutes by a technician with minimum training in chemistry.
Analysis of leaf samples - Results from the analysis of the three samples of Erythroxylum coca Lam. are summarized in table 3. The cocaine content (0.57 to 0.60 per cent) in these three samples was comparable to those reported for Bolivian and Java coca leaves [ 1] . It must be mentioned that other alkaloids were detected in the analysed extracts which had short retention times and thus appeared with the solvent front. Since our primary concern in this publication was the cocaine content, no attempt was made to reduce the column temperature and analyse for the other alkaloids. This will be the subject of future communications.
|
Time (minutes) |
Cocaine determined a (percentages) |
Coefficient of variation (percentages) |
|---|---|---|
| 15 |
0.60 ± 0.03
|
5.0 |
| 30 |
0.65 ± 0.09
|
14.0 |
| 45 |
0.54 ± 0.07
|
13.0 |
| 60 |
0.57 ± 0.09
|
16.0 |
aMean of triplicate determinations.
|
Species |
Observed concentration a (percentages) |
Coefficient of variation b (percentages) |
|
|---|---|---|---|
|
Cuzco
|
0.60 | 0.03 | 5.0 |
|
Trujillo
|
0.60 | 0.03 | 5.0 |
|
Tingo Maria
|
0.57 | 0.01 | 1.8 |
aMean of triplicate determination.
bMean coefficient of variation=3.9 per cent.
S. Archer and R.L. Hawks in Cocaine: Chemical, Biological, Clinical, Social and Treatment Aspects , S.J. Mulé, ed. CRC Press, Cleveland, Ohio, p. 16 (1976).
002G.H. Aynilian, J. A. Duke, W. A. Gentner and N. R. Farnsworth,; J. Pharm. Sci., 63 , 1938 (1974).
003B. Holmstedt, E. Jaatmaa, K. Leander and T. Plowman; Phytochemistry, 16 , 1753 (1977).
004M. L. Bastos and D.B. Hoffman in Cocaine: Chemical, Biological, Clinical, Social and Treatment Aspects , S. J. Mulé, ed. CRC Press, Cleveland, Ohio, pp. 35-38 (1976).
005P. Jatlow, ibid., pp. 61-69.
006J.E. Wallace, H.E. Hamilton, D.E. King, D. J. Bason, H.A. Schwertner and S.C. Harris. Anal. Chem ., 48 , 34 (1976).
007B.H. Duorchik, S.H. Miller and W. P. Graham. J. Chromatogr., 135 , 141 (1977).
008D.L. Minden and N. A. D. Amato, Anal. Chem. 49 , 1974 (1977).
009M.J. Kogan, K.G. Verebey, A.C. DePace, R.B. Resnik and S.J. Mulé, ibid., (1965).
010S.P. Jindal and P. Vestergaard. J. Pharm. Sci., 67 , 811 (1978).
011J.I. Javad, H. Dekirmenjian, J.M. Davis and C.R. Schuster. J. Chromatogr., 152 , 105 (1978).
012J.C. Valentour, V. Aggarwal, M.P. McGee and S.W. Goza. J. Anal. Toxicol., 2 , 134 (1978).


