ABSTRACT
Introduction
Material and methods
Results and discussion
Acknowledgement
References
Author: G. SINISCALCO GIGLIANO, A. DI FINIZIO
Creation Date: 1999/12/01
G. SINISCALCO GIGLIANO
A. DI FINIZIO
Dipartimento di Biologia Vegetale, Università di Napoli Federico II,
via Foria 223, 80139 Naples, Italy
The authors report on a method to identify unknown samples of plant material as Cannabis sativa L. The method involves polymerase chain reaction (PCR) amplification of the internal transcribed spacer I (ITS1) of the nuclear ribosomal deoxyribonucleic acid (n-rDNA) in five different accessions of C. sativa from various geographical areas, as well as in one accession of Humulus lupulus L., which belongs to the only other genus of the family Cannabaceae.
The use of ITS1, amplified and successively digested with appropriate restriction endonucleases, has allowed the construction of a Cannabis fingerprint that can be used in forensic investigations for the identification of samples suspected of being Cannabis.
The method reported in the present paper has the merit of making it possible to process very small amounts of material; it is also not expensive, does not require access to a sequencing facility and does not utilize sophisticated apparatus.
Botanical identification of Cannabis material for forensic purposes is normally carried out by microscope examination and concentrates on leafy material. The type and nature of cystolith hairs on the leaves, as well as the cellular structure of the seeds, are examined.
Chemical identification of cannabinoids, on the other hand, can be carried out by using a variety of different methods (thin-layer chromatography, gas chromatography or gas chromatography/mass spectrometry).
The techniques mentioned above, although working fairly well in certain cases, show severe limitations, especially when the experimental material is poorly preserved. For example, as far as botanical investigation is involved, Nakamura [1] described more than 80 different plant species containing cystolith hairs similar to those found in Cannabis. It is thus conceivable that suspicious plant material could be erroneously identified as Cannabis.
Another problem is related to chemical testing, namely, that the cannabinoids and especially the tetrahydrocannabinol (THC) are readily oxidized [2] and therefore the absence of detectable THC in an unidentified sample does not prove that it is not marijuana.
The task of the expert in charge of forensic investigations can be particularly arduous when the plant material under study has been previously treated (e.g. minced, desiccated or macerated), has been poorly stored by the criminals or by the police after seizure or has been seized in very small amounts. In these extreme cases, unfortunately rather common in forensic practice, identification, as well as testing for the presence of cannabinoids, can be almost impossible. For these reasons, a method allowing plant identification in a way almost completely independent of the quantity of starting material and its state of preservation would be extremely useful to the forensic expert.
Such methods can be developed and the description of one such method is the object of the present paper. The authors would like to stress that this type of method allows identification of unknown plant material as C. sativa, but cannot give answers about content in cannabinoids. Such methods are nevertheless forensically relevant, as in different countries on various continents (e.g. Denmark, Colombia and the United Republic of Tanzania) cultivation of C. sativa is forbidden per se, regardless of the content in cannabinoids.
In recent years, the study of deoxyribonucleic acid (DNA) has steadily gained in interest for the general forensic expert, especially in terms of identifying criminals by using traces of their body fluids in cases of violent crime with no witnesses. The new techniques rely on an advance of paramount importance in the field of molecular biology, the polymerase chain reaction (PCR). By using this method, at present comparatively simple and inexpensive, it is possible to obtain large amounts of a given small fragment of DNA (i.e. to amplify a specific DNA sequence) starting from minimal quantities of total DNA, by using specific, very small DNA fragments as primers. Total DNA can be extracted from very small amounts of tissue or body fluid of any organism (even in cases where a long time has elapsed and preservation has been poor). For example, by using PCR techniques molecular biologists have succeeded in extracting DNA from plant fossils [3] and from human mummies [4]. If the object of the reaction is an individual-specific DNA sequence, a single individual can be identified (provided that a sample of his/her DNA can be obtained for comparison); if the PCR has a species-specific DNA fragment as its target, the unidentified starting material can be attributed to a given biological species with certainty, provided that a reference DNA sequence is known for that species.
Once the DNA fragment has been obtained by means of PCR, various strategies can be pursued in order to compare it with a reference DNA. The best way is to sequence the fragment, that is, to obtain information on the physical succession of nucleotides (the monomers of DNA) in the fragment. Sequencing requires a fully equipped molecular biology laboratory, reagents that at present are still rather expensive (especially to obtain in developing countries) and handling of radioisotopes (with all the expense and safety precautions involved), or, as an alternative to the latter, the purchase of an automated sequencing apparatus, which is still a very expensive piece of equipment.
One of the authors of the present paper has already developed such a method in order to identify unknown plant material as C. sativa [5]. The method uses a sequence of ribosomal DNA (i.e. the DNA that codes for the ribonucleic acid (RNA) component of ribosomes).
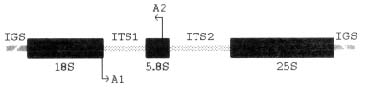
The sequence of fragments of such DNA is known for a large number of species. In plants the ribosomal DNA is made of repeated units present in a few hundreds to more than tens of thousands of copies in each nucleus [6]. Each unit consists of three subunits (18S, 5.8S and 25S), which code for the ribosome itself. These regions are separated by two quite variable internal transcribed spacers (ITS1 and ITS2). Units are in turn separated by a highly variable inter-genic spacer (IGS) (see figure I).
Figure I. Organization of ribosomal genes: A1 and A2 indicate the anneal region of the primers for the ITS1 amplification
In the above-mentioned paper [5], the sequence of ITS2 was examined for five different strains of C. sativa, showing that ITS2 was almost invariant among Cannabis strains and different from that of other plant species. Therefore, by obtaining the ITS2 sequence from an unidentified sample of plant material and by comparing it with the reference sequence of Cannabis, it is possible to identify the sample as Cannabis.
The object of this paper is to test the feasibility of using ITS1, the other internal spacer of ribosomal DNA, in the construction of Cannabis "fingerprints" and the comparison of it with a sample suspected of being Cannabis. The choice of this region (ITS1) is also based on a previous paper [7], in which the authors have shown that ITS1 is homogeneous in length in different Cannabis accessions. However, the technique used to compare the reference sample with the unknown ITS1 obtained is different from that described in Siniscalco Gigliano et al. (1997) [5]. Discrimination relies in this case on the use of specific enzymes (restriction endonucleases) with which the investigated DNA is digested. Restriction endonucleases (discussed further in the relevant section under materials and methods) cleave the digested DNA into a set of specific fragments, which can be visualized and used for comparison. By employing various different endonucleases in different reactions, it is possible to obtain various groups of specific DNA fragments that together allow the identification of a sample. By increasing the number of endonucleases employed, a level of accuracy can be achieved that is only slightly inferior to the absolute certainty given by the sequencing approach.
The method described here, while slightly less accurate than sequencing, has been devised for specific purposes: it can be completed in a shorter time; it does not require very sophisticated apparatus (the most expensive single item being the PCR thermocycler); it has an overall cost roughly one order of magnitude less than sequencing; and it requires a shorter period of training. This method, which is most suitable in situations in which sequencing facilities are not readily available, can also be used as a quick screening method when a large number of samples must be processed. (In fact, the lower accuracy mentioned above relates to positive identification; negative identification, that is, stating that an unknown sample is not Cannabis, is on the contrary as certain as with sequencing.)
Plant material
Five different Cannabis accessions were used, plus one accession of Humulus lupulus L., which belongs to the only other genus of the family Cannabaceae. The list of accessions used (names and geographical origins) is given in table 1.
Table 1. Names, accession numbers and geographical origins
of the
Cannabis and
Humulus cultivars used
| Taxon | Accession number * | Geographic origin |
| Cannabis sativa var. indica | CJBN 716/85 | France |
| Cannabis indica | CPRO-dlo 883271 | Afghanistan |
| Cannabis | CPRO-dlo 891191 | Nepal |
| Cannabis | SS 241 | The Netherlands |
| Cannabis sativa | OBN 0148-F | Italy |
| Humulus lupulus | OBN 2801-F | Italy |
a CJBN Conservatoire et Jardins botaniques, Nancy, France
CPRO-DLO Centre for Plant Breeding and Reproduction Research, Wageningen, The Netherlands
SS Sensi Seeds, Rotterdam, The Netherlands
OBN Orto botanico, Università di Napoli, Italy
DNA extraction
DNA was extracted from dried (0.05-1 g) or fresh (0.1-1 g) leaves. Extraction was carried out by using a protocol described by Caputo et al. [8], appropriately scaled and modified. Samples were ground in liquid nitrogen in a small mortar and carefully transferred to a 1.5 ml disposable microcentrifuge tube, paying attention not to exceed a volume of approximately 300 el. Immediately after the nitrogen evaporation, 800 el of extraction buffer (50 mM Tris-HCl pH 8.0; 20 mM EDTA pH 8.0; 0.2 per cent bovine serum albumine; 1 per cent polyvinilpirrolidone; and 0.1 per cent β-mercaptoethanol) was added to the tissue powder. Cells were lysed by adding sodium dodecyl sulphate and sodium N-lauroylsarcosinate to a final concentration of 2 per cent each and incubated for 15 minutes in a water bath set at 67° C. Samples were briefly cooled in an ice bath and proteins were precipitated by adding 0.3 vol 5M potassium acetate, followed by a 20-minute incubation on ice and a 20-minute centrifugation in an Eppendorf microfuge at maximum speed (approx. 14,000 x g) at 4° C. The supernatant was extracted twice or three times with chloroform-isoamyl alcohol (24:1) and DNA precipitated by adding 2 vol ethanol and 0.1 vol 3M sodium acetate. The samples were briefly frozen in an ultrafreezer and then centrifuged for 15 minutes under the same conditions as above. The pellet was then resuspended in approximately 500 µl of redistilled water. DNA was precipitated again with 1/9 5M NaCl and 20 per cent polyethylene glycol (PEG-8000) (equal volume). Vials were then frozen in liquid nitrogen and stored at -80° C for 30 minutes. Finally, the DNA precipitate was collected by centrifugation for 15 minutes (approx. 14,000 x g at 4° C), washed again in 70 per cent ethanol and resuspended in a suitable volume of redistilled water.
DNA amplification
The ITS1 region was amplified by PCR using two primers, one of which annealed in the 3' region of the 18S (5'-GGAGAAGTCGTAACAAGGTTTCCG-3') and the other in the 5' region of the 5.8S (5'-ATCCTGCAATTCACACCAAGTATCG-3') rDNA genes, respectively (see figure I).
PCR reactions were conducted in a thermal cycler Cetus 9600 (Perkin Elmer) for 30 cycles.
The final volume of the PCR mixture was 100 µl and consisted of a 2-10 ng DNA sample, 10 µl of buffer (500 mM KCl, 100 mM Tris-HCl pH 9.0, 1 per cent Triton X-100 and 25 mM MgCl2), 1 µl of primer (0.25 M), 0.2 mM each of the four dNTPs and 2.5 units of Taq polymerase.
Initial conditions were as follows: one-minute denaturation at 94° C, one-minute annealing at 55° C and 45-second extension at 72° C. Samples were denatured for five minutes at 94° C before the beginning of the first cycle; extension time was increased by three seconds per cycle; and extension was further prolonged for seven minutes at the end of the last cycle.
PCR products were then column-purified by using Microcon 100 micro-concentrators (Amicon, Cat. 42413) and loaded onto a 2-per-cent agarose gel and electrophoresed under the same conditions as reported below.
Digestion of restriction endonucleases
A large number of restriction endonucleases were evaluated. Restriction endonucleases are enzymes that cleave DNA if, and only if, they recognize a particular sequence of four or more bases on the DNA sequence (see table 2). Such enzymes can be readily purchased for various manufacturers and several hundreds are available that recognize different DNA sequences, commonly 4 or 6 base pairs (bp) long. The final selection was made so as to choose endonucleases that produced fragments that could be clearly visualized in the authors' gel system. The restriction endonucleases tested, which produced characteristic fragments, are reported in table 2.
Table 2. Restriction endonucleases tested that produced characteristic fragments
in ITS1 of
Cannabis and
Humulus: cleavage sequence per enzyme
| Enzyme | Isoschizomer | Specificity* |
| Ava I | Eco 88I | C|YCGRG |
| Ava II | G|GWCC | |
| Bst NI | Mva I | G|AATTG |
| Cla I | Bsu 15I | AT|CGAT |
| Eco RII | |CCWGG | |
| Eco RV | Eco 32I | GAT|ATC |
| Esp 3I | CGTCTCN| | |
| Fnu DII | Mvn I | CG|CG |
| Fnu 4HI | Ita I | GC|NGC |
| Hae III | GG|CC | |
| Hgi AI | Asp HI | GWGCW|C |
| Hinf I | G|ANTC | |
| Hpa II | C|CGG | |
| Mae I | C|TAG | |
| Mbo II | GAAGAN8| | |
| Nla IV | Bsp LI | GGN|NCC |
| Sdu I | GDGCH|C | |
| Xho I | C|TCGAG |
a D = A, G or T; H = A, C or T; N = A, G, C or T; R = A or G; W = A or T; and Y = C or T.
PCR-purified fragments were then digested with the selected restriction endonucleases according to the manufacturer's specifications.
Electrophoresis and agarose gel
Digested samples were loaded onto a 2-per-cent agarose gel (Boehringer type MP) prepared in a 1xTBE buffer (0.09 M Tris-borate and 0.002 M EDTA pH 8.0) containing 50 ng/ml ethidium bromide and electrophoresed at 9 V/cm. The length of the fragments was estimated by using a 100-bp DNA ladder (Promega, Cat. G3161) as marker.
The DNA bands were visualized using an ultraviolet light transilluminator (254 nm) and photographed using 667 Polaroid film.
The Cannabis and Humulus ITS1 DNA fragments obtained by PCR are shown in figure II.
Figure II. Cannabis and Humulus ITS1 DNA fragments obtained by PCR a
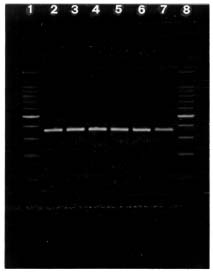
a Lanes 2-6, Cannabis; lane 7, Humulus; and lanes 1 and 8, 100-bp DNA ladder (the bright fragment is 500 bp long).
Table 3 shows a list of all the restriction endonucleases tested that produced characteristic fragments. The number of cuts and length of fragments produced are reported for each enzyme.
Table 3. Restriction endonucleases tested that produced characteristic
fragments in ITS1 of
Cannabis and
Humulus: number of cuts
and length of fragments produced per enzyme
| Number of cuts | Length of fragments produced (bp) | |||
| Enzyme | Cannabis | Humulus | Cannabis | Humulus |
| Ava I | 0 | 1 | 360 | 250 - 110 |
| Ava II | 1 | 1 | 130 - 230 | 130 - 230 |
| Bst NI | 0 | 1 | 360 | 160 - 200 |
| Cla I | 1 | 1 | 300 - 60 | 300 - 60 |
| Eco RII | 0 | 1 | 360 | 160 - 200 |
| Eco RV | 1 | 1 | 300 - 60 | 300 - 60 |
| Esp 3I | 1 | 1 | 230 - 130 | 230 - 130 |
| Fnu DII | 1 | 1 | 210 - 150 | 210 - 150 |
| Fnu 4HI | 0 | 1 | 360 | 235 - 125 |
| Hae III | 1 | 0 | 220 - 140 | 360 |
| Hgi AI | 0 | 1 | 360 | 250 - 110 |
| Hinf I | 1 | 1 | 280 - 80 | 280 - 80 |
| Hpa II | 2 | 2 | 180 - 60 - 120 | 180 - 60 - 120 |
| Mae I | 0 | 1 | 360 | 150 - 210 |
| Mbo II | 0 | 1 | 360 | 260 - 100 |
| Nla IV | 1 | 1 | 130 - 230 | 130 - 230 |
| Sdu I | 0 | 1 | 360 | 250 - 110 |
| Xho I | 0 | 1 | 360 | 250 - 110 |
The fragments obtained after digestion with the same representative restriction endonucleases tested are shown in figure III.
The results obtained show that the ITS1 length is homogeneous in the five different accessions of Cannabis and in Humulus (360 bp). As external primers were used, each fragment includes the ITS1 region plus an upstream fragment relative to the 3' region of the 18S and a downstream fragment relative to the 5' region of the 5.8S.
Figure III.
Cannabis and Humulus ITS1 DNA fragments obtained
by PCR and digested with the most representative restriction
endonucleases tested
a

a Cannabis (lane 2) and Humulus (lane 3),digested with Ava I; Cannabis (lane 4) and Humulus (lane 5), digested with Fnu 4HI; Cannabis (lane 6) and Humulus (lane 7), digested with Hae III; and Cannabis (lane 8) and Humulus (lane 9),digested with Xho I. Lanes 1 and 10, 100-bp DNA ladder (the bright fragment is 500 bp long).
However, following comparison of the angiosperm sequences of the 18S and 5.8S available in the literature and consulted by accessing the GenBank database, the upstream (50 bp) and downstream (85 bp) fragments of the ITS1 were excluded from the length of the amplified regions. For this reason, the ITS1 of the taxa in the study are approximately 225 bp long.
The restriction endonucleases Ava I, Bst NI, Eco RII, Fnu 4HI, Hgi AI, Mae I, Mbo II, Sdu I and Xho I produced fragments in the ITS1 of Humulus but not in that of Cannabis, while Hae III is the only restriction endonuclease that produced fragments in the ITS1 of Cannabis but not in Humulus.
The restriction endonucleases Ava II, Cla I, Eco RV, Esp 3I, Fnu DII, Hinf I, Hpa II and Nla IV produced fragments of the same length in Cannabis and in Humulus. All the five accessions of Cannabis produced the same fragments after digestion with the restriction endonucleases tested. Figure III shows the fragments obtained for one of the Cannabis accessions tested with the most representative enzymes used.
The two fragments produced in Humulus by using Ava I, Fnu 4HI and Xho I are 250 and 110 bp, 235 and 125 bp and 250 and 110 bp, respectively. The two fragments produced in Cannabis from Hae III are 220 and 140 bp.
The results show that ITS1 is an ideal molecule to construct a restriction map of Cannabis DNA that can be used in forensic investigations for the identification of a sample suspected of being Cannabis.
The method reported in this paper, as that described in a previous paper [5], has the merit of making it possible to process very small amounts of material (as little as 50 mg of dried material); it is also not expensive, does not require access to a sequencing facility and does not utilize sophisticated apparatus.
We acknowledge the courtesy of Dr. Loek J. M. Soest (Centre for Plant Breeding and Reproduction Research, The Netherlands) in providing hemp seeds.
1. G. R. Nakamura, "Forensic aspects of cystolith hairs of Cannabis and other plants", Journal of AOAC International, vol. 52 (1969), pp. 5-16.
2. R. Gillan and others, "Comparison of Cannabis sativa by random amplification of polymorphic DNA (RAPD) and HPLC of cannabinoids: a preliminary study", Science & Justice, vol. 35 (1995), pp. 169-177.
3. E. M. Golenberg and others, "Chloroplast DNA sequence from a Miocene magnolia species", Nature, vol. 344 (1990), pp. 656-658.
4. S. R. Woodward and others, "Amplification of ancient nuclear DNA from teeth and soft tissues", PCR Methods Application, vol. 3 (1994), pp. 244-247.
5. G. Siniscalco Gigliano, P. Caputo and S. Cozzolino, "Ribosomal DNA analysis as a tool to identify specimens of Cannabis sativa L. of forensic interest", Science & Justice, vol. 37 (1997), pp. 171-174.
6. R. Appels and R. L. Honeycut, "rDNA: evolution over a billion years", DNA systematics, S. K. Dutta, ed. (Boca Raton, FL., CRC Press, 1986), vol. II: Plants, pp. 81-135.
7. G. Siniscalco Gigliano and A. Di Finizio, "Approccio molecolare nelle indagini forensi su Cannabis sativa L.", Delpinoa, vol. 36 (1994), pp. 15-28.
8. P. Caputo, D. W. Stevenson and E. T. Wurtzel, "A phylogenetic analysis of American Zamiaceae ( Cycadales) using chloroplast DNA restriction fragment length polymorphisms", Brittonia, vol. 73 (1991), pp. 135-145.
|
|


